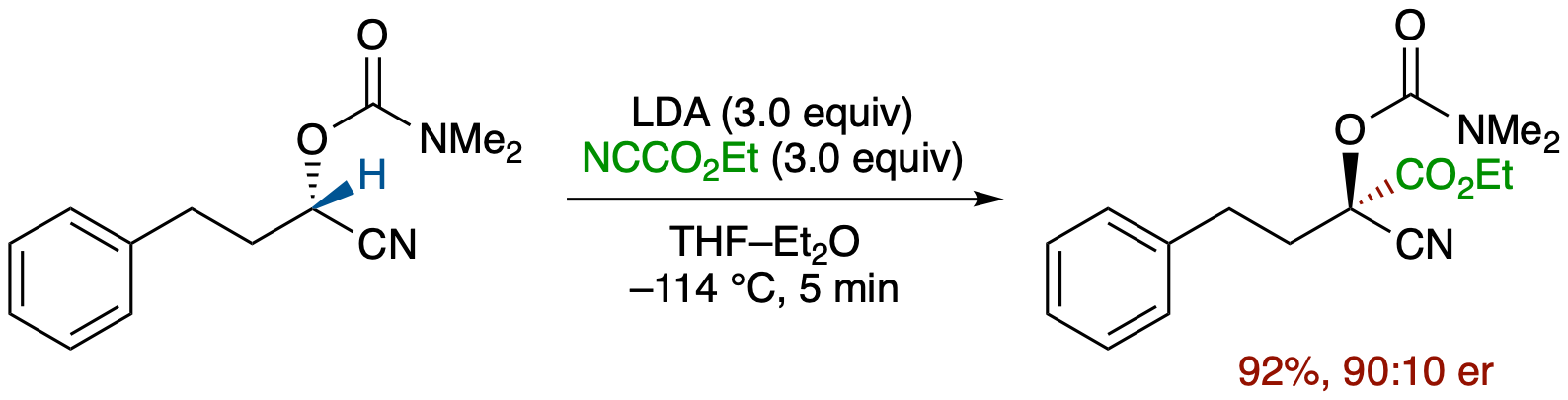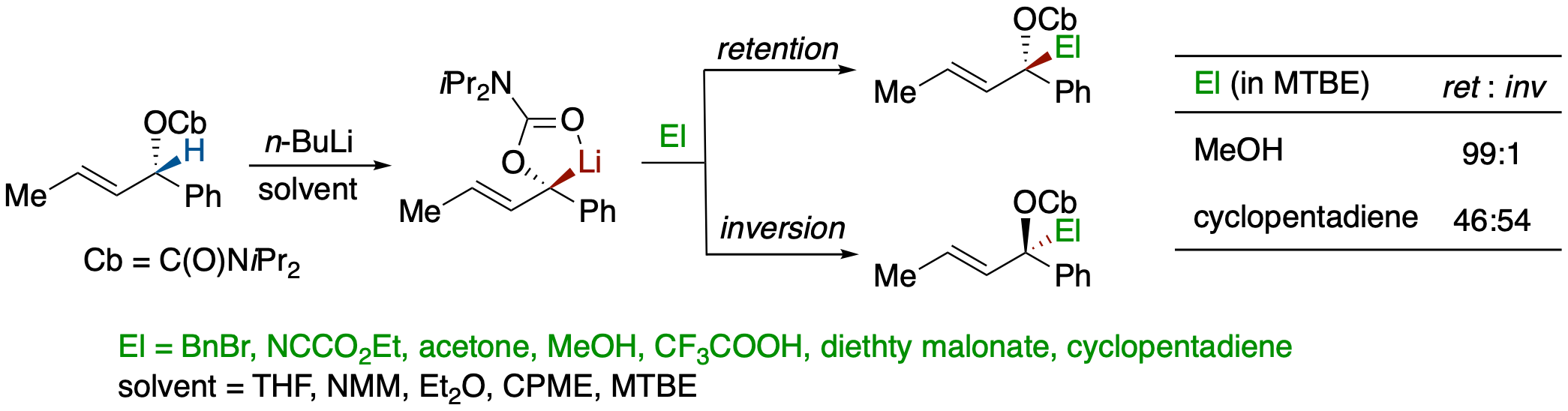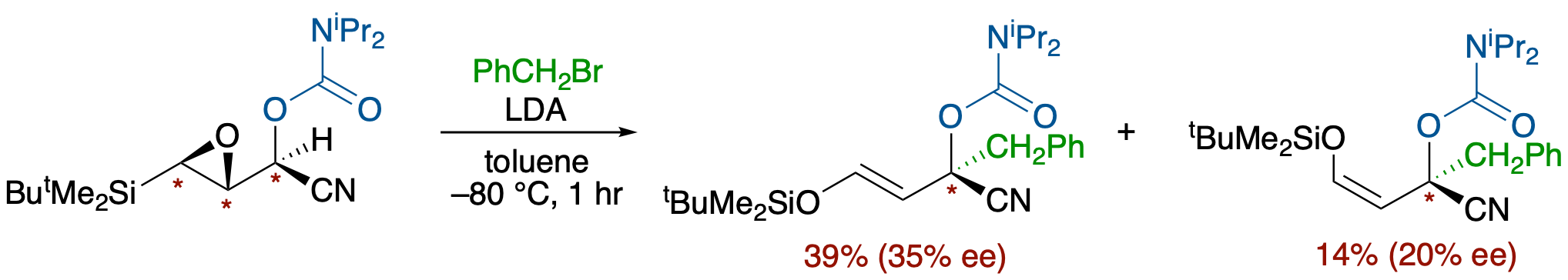Research
Enantioselective Cyanation-Brook Rearrangement-Protonation of Acylsilanes and Stereospecific Inversion in Brook Rearrangements Generating α-Nitrile Carbanion Equivalents
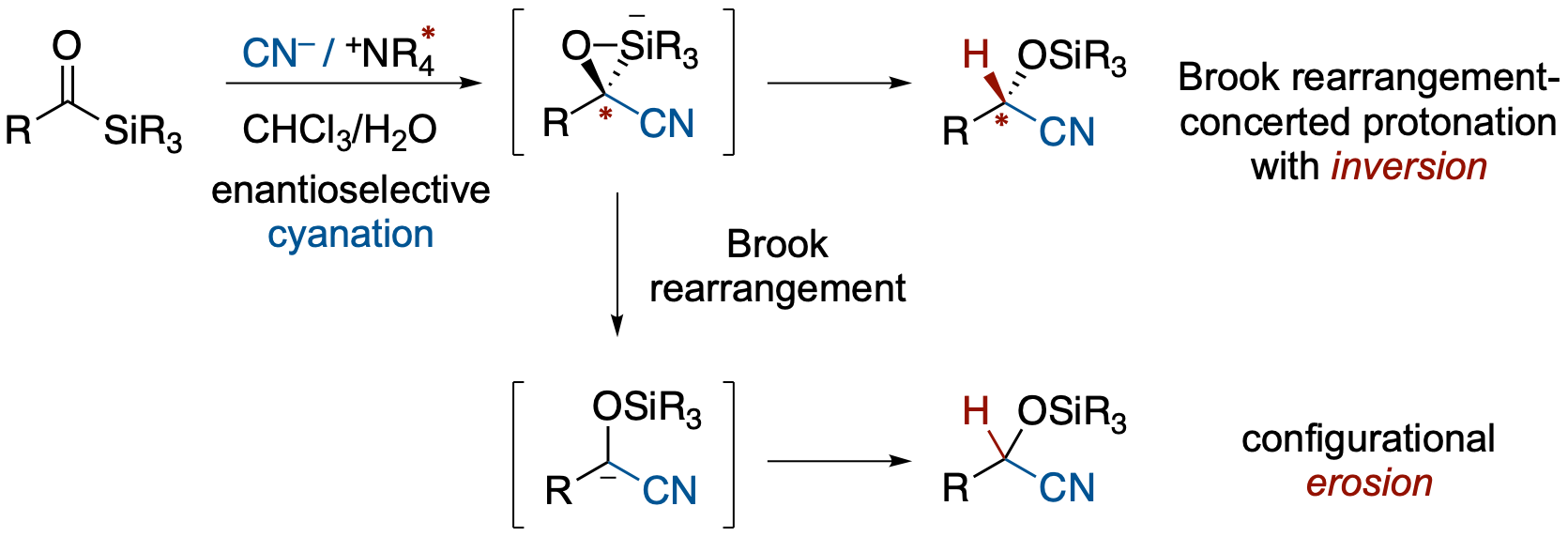
- Chiral α-nitrile carbanions are attractive intermediates in organic synthesis, but their use in asymmetric reactions has been limited because they readily lose configurational integrity. We have developed an enantioselective cyanation–Brook rearrangement–protonation sequence of acylsilanes under phase-transfer catalytic conditions, providing enantioenriched O-silyl cyanohydrins in good yields and up to 92:8 er. Mechanistic studies using independently prepared enantioenriched α-silyl cyanohydrins revealed that the Brook rearrangement generating a formal α-nitrile carbanion equivalent proceeds with stereospecific inversion of configuration. The overall enantioselectivity is introduced mainly during the initial cyanation step, whereas the subsequent Brook rearrangement–protonation sequence determines how much of the stereochemical information is retained or eroded. Countercation effects, crossover experiments, and isotope-effect studies support a mechanistic picture in which stereochemical fidelity is governed by competition between concerted protonation within a silicate ion pair and stepwise pathways involving delocalized α-nitrile anionic character. These findings establish cyanation–Brook rearrangement–protonation as a useful platform for both asymmetric synthesis and the mechanistic study of short-lived, configurationally labile carbanion equivalents.
Abnormal Michael Reaction: Conditions-Dependent Product Distributions in the Michael Reaction of Monoalkyl-Substituted Malonates
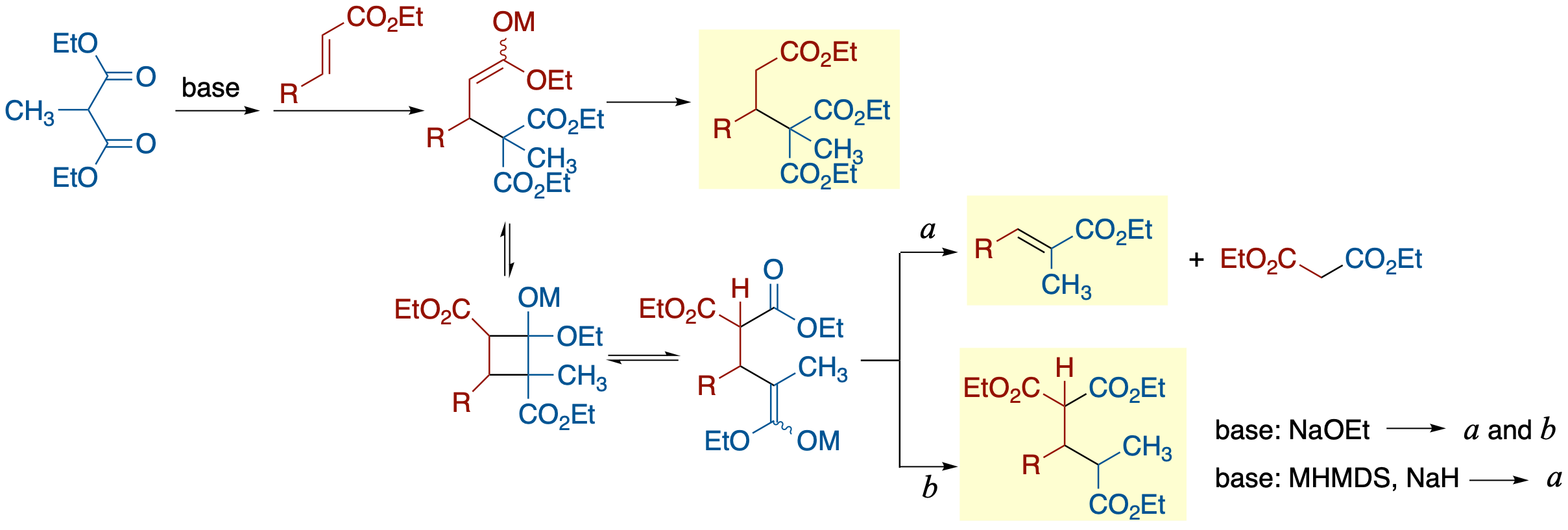
- The abnormal Michael reaction reported by Thorpe and Michael in 1900, which involves the transfer of an activating group from the nucleophilic species to the α-carbon of the Michael acceptor in reactions with monosubstituted malonates, was revisited using prototypical substrates for both intramolecular and intermolecular reactions. In both reactions, conditions-dependent product distributions were observed. Thus, under the conditions using NaHMDS or NaH in THF or Et2O, no formation of the abnormal Michael product was observed, and the major product was an apparent retrograde Michael reaction in the abnormal Michael product resulting from an elimination of a malonate anion with the migrated acyl group as a part of it. The use of a base capable of generating a proton source such as NaOEt resulted in formation of the abnormal Michael product in the intermolecular reaction, although the retrograde Michael product was still a major product. On the other hand, in the intramolecular reaction, the abnormal product was not detected under any conditions used. A plausible reaction mechanism in which the lower kinetic acidity of the malonate proton compared to the thermodynamic acidity plays a significant role has been proposed.
Steric Course of Deprotonation/Substitution of Chelating/Dipole-Stabilizing-Group-Substituted α-Amino- and α-Oxynitriles
- The stereochemical course of electrophilic substitutions of α-nitrile metallocarbanions was investigated using deprotonation of enantioenriched α-amino- and α-oxynitriles bearing a carbamoyl or a methoxycarbonyl group in the presence of an electrophile. The results suggested that the dipole-stabilizing substituents can not only affect the configurational stability and chemical reactivity of α-nitrile metallocarbanions but also can change the steric course of the electrophilic substitution (retention vs inversion). While the reaction of α-aminonitrile bearing a dimethylaminocarbonyl group on the nitrogen atom with LiHMDS in the presence of PhCOCl resulted in the formation of a retention product (93:7), the case with that bearing a methoxycarbonyl group was reversed in favor of the inversion product (37:63). Thus, a methoxycarbonylamino group increases the degree of planarization of the anionic center with maintenance of the planar chirality more than does a ureido group, leading to preferable attack of an electrophile from the rear face.

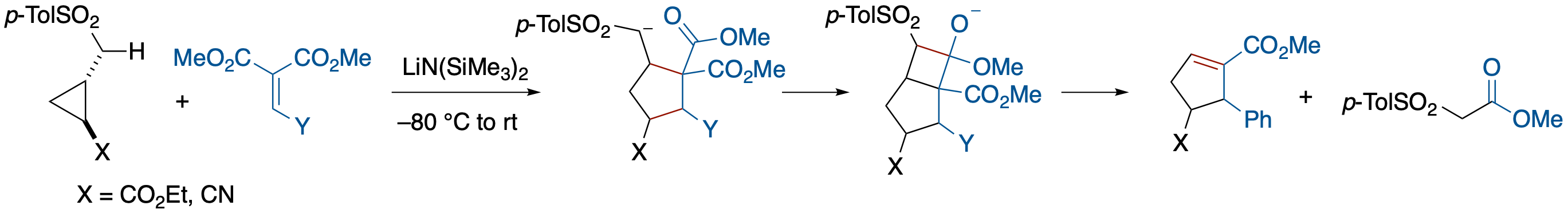
Carbanion-Induced [3 + 2]-Annulation of Donor-Acceptor Cyclopropanes
- [3 + 2]-annulation of donor-acceptor cyclopropanes and ylidenemalonates in which an α-p-tosyl carbanion functions as a donor substituent is described. A notable feature of the annulation is that the auxiliary p-tosylmethyl group can be removed via a cycloreversion during the tandem annulation sequence.
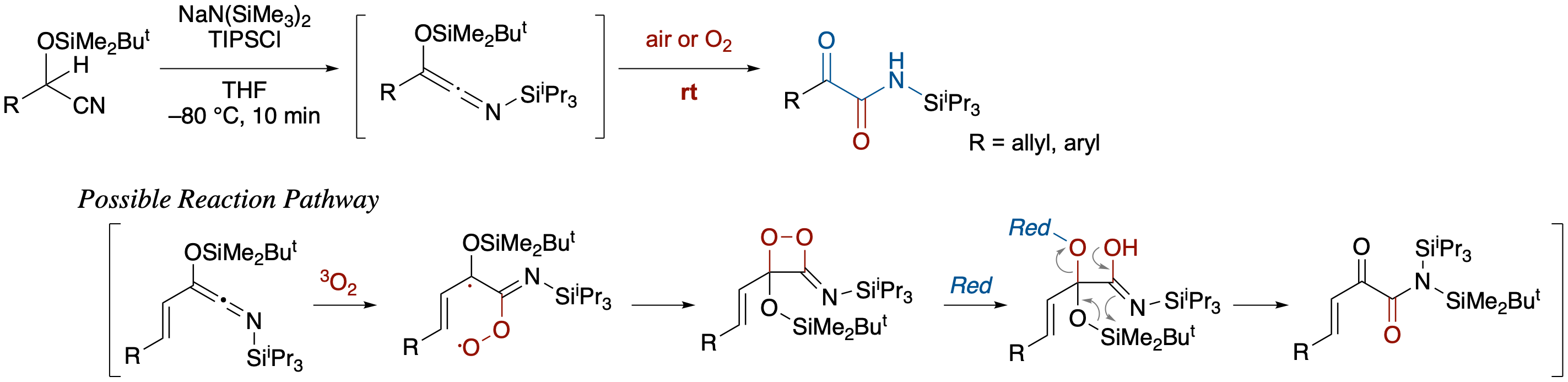
Spontaneous Oxygenation of Siloxy-N-Silylketenimines to α-Ketoamides
- Siloxy-N-silylketenimines generated in situ from O-silyl cyanohydrins were converted to α-ketoamides by brief exposure to air or oxygen. Oxidation under extremely mild conditions can be explained by assuming the intermediacy of a 3-imino-1,2-dioxetane derivative generated via triplet-singlet intersystem crossing after the reaction of siloxy-N-silylketenimines with triplet oxygen.
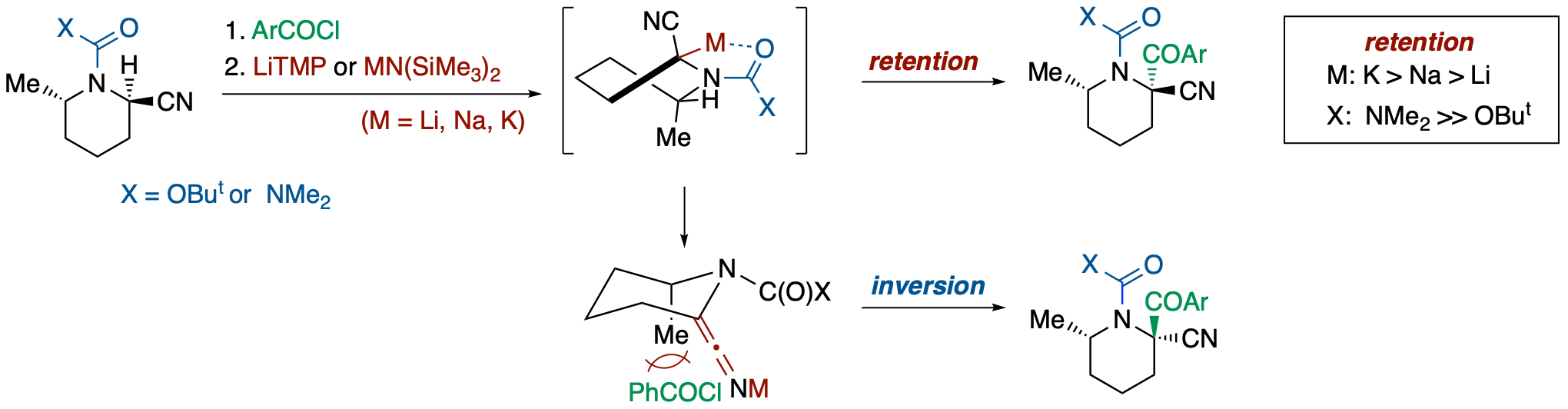
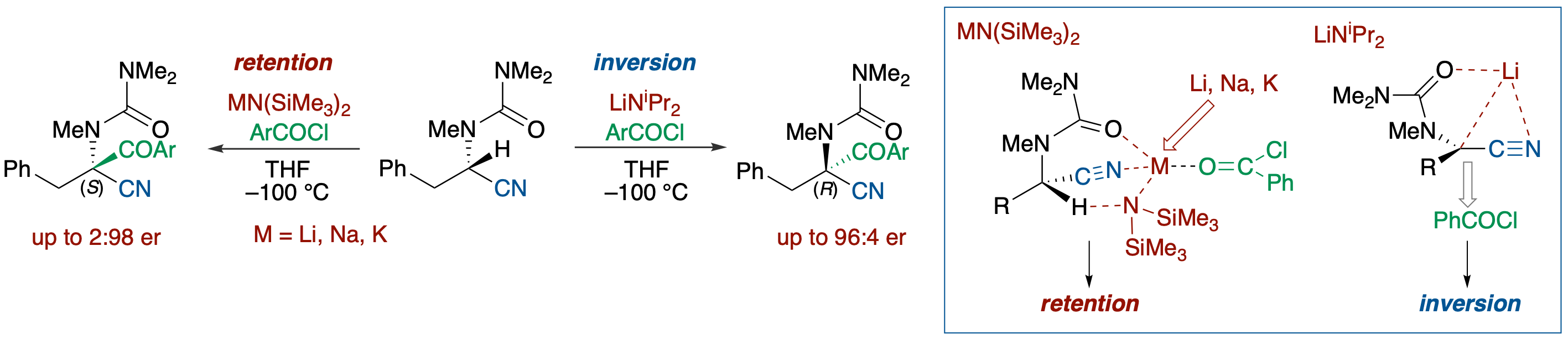
Stereochemical Course of Deprotonation-Acylation of N‐Boc- and N‐Carbamoyl-2-cyano-6-methylpiperidines
- The stereochemical course of electrophilic substitution of α-nitrile metallocarbanions generated by deprotonation from N-Boc- and N-carbamoyl-2-cyano-6-methylpiperidines was investigated. Deprotonation in the presence of an electrophile taking advantage of the high acidity of α-nitrile protons allowed examination of the effects of a chelating group on the nitrogen atom, a countercation, and the reactivity of an electrophile on the steric course. Analyses of reactions using aroyl chlorides and methyl iodide revealed the following: : (1) the substitution reactions basically proceed with retention of configuration, (2) the extent of an inversion product increases with decreasing chelating ability of the N-substituent and with increasing leaving ability (ionic character) of a countercation (Li, Na, K) of the anionic species, and (3) the use of a more reactive electrophile results in an increase of the retention product.
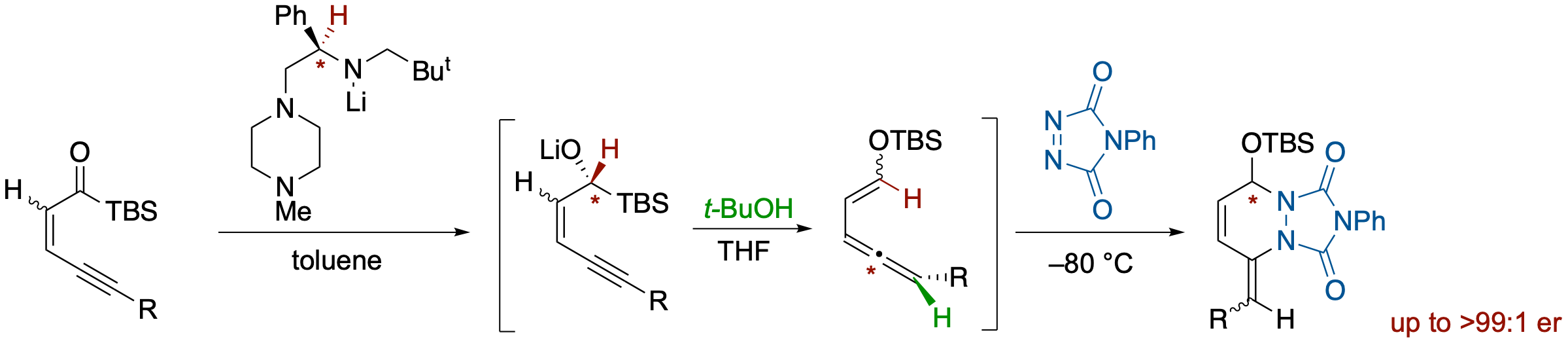
Enantioselective Synthesis of Allenylenol Silyl Ethers via Chiral Lithium Amide-Mediated Reduction of Ynenoyl Silanes and Their Diels-Alder Reactions
- An enantioselective Meerwein-Ponndorf-Verley-type reduction of ynenoylsilanes by a chiral lithium amide followed by a Brook rearrangement and anti-mode protonation across conjugated 1,3-enynes provides allene derivatives bearing a 2-siloxyvinyl moiety in high enantioselectivity. The E/Z geometry of enol silyl ethers is controlled by the geometry of the starting enyne moiety. Thus, (E)- and (Z)-enol silyl ethers are obtained from (Z)- and (E)-ynenoylsilans, respectively. The 2-siloxyvinylallene products can participate in Diels-Alder reactions with reactive dienophiles such as PTAD, which can be achieved in a one-pot operation from ynenoylsilanes.
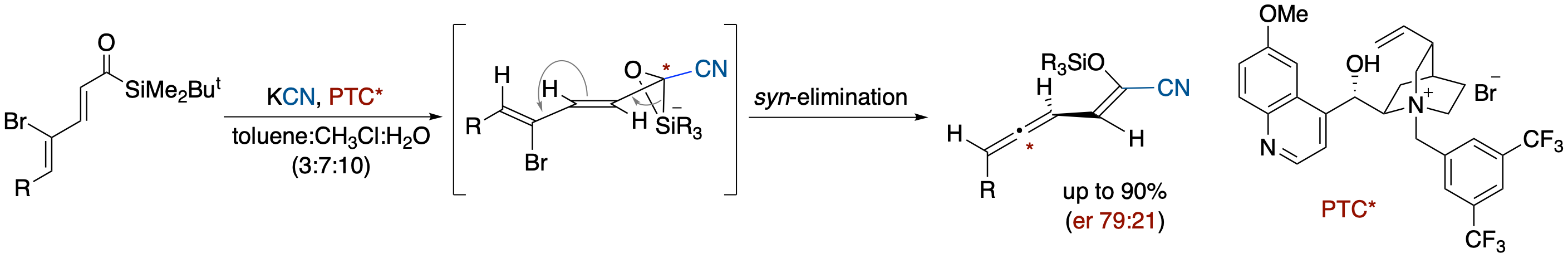
Formation of 2-Cyano-2-siloxyvinylallenes via Cyanide-Induced Brook Rearrangement in γ-Bromo-α,β,γ,δ-unsaturated Acylsilanes
- Reactions of γ-bromo-α,β,γ,δ-unsaturated acylsilanes with KCN under phase-transfer catalyst conditions using n-Bu4NCN afforded 2-cyano-2-siloxyvinylallenes via a tandem process that involves a nucleophilic attack of a cyanide ion and a Brook rearrangement-induced conjugate vinylic 1,4-elimination. Use of a chiral cyanide ion source, derived from KCN and quaternary ammonium bromide derived from cinchona alkaloids, provided nonracemic allene derivatives. Based on this result and the reaction using a chiral hydride ion source, we propose a reaction pathway in which a Brook rearrangement-mediated vinylic conjugate 1,4-elimination occurs in a syn alignment between the C-Br bond and C-Si bond in the silicate intermediate.
Enantioselective Synthesis of a-Silylamines by Meerwein-Ponndorf-Verley-Type Reduction of
α-Silylimines by a Chiral Lithium Amide
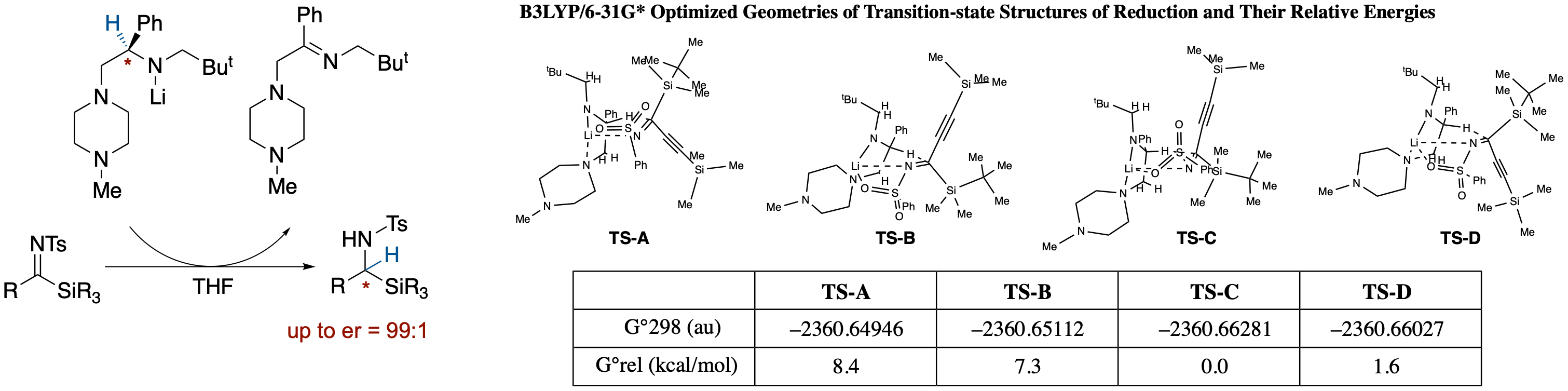
- Meerwein-Ponndorf-Verley-type reduction of N-tosylsilylimines with chiral lithium amide affords α-silylamines in high enantioselectivity. Since the enantioselectivity observed was inconsistent with our previously proposed chair-like six-membered transition structure, we performed density functional theory (DFT) calculations on transition states using an N-phenylsulfonyl derivative as a model system. Results of the calculations showed that the structures are considerably deformed from the chair-like form with steric repulsions between the 1'-methylene group and the imine-carbon substituents playing an important role in the control of the enantioselectivity.
Enantiodivergent Deprotonation-Acylation of α-Amino Nitriles
- Deprotonation-acylation of enantioenriched α-ureidonitriles, readily derived from α-amino acids, was found to proceed in a highly enantiodivergent manner despite the required intermediacy of extremely stereolabile α-nitrile metalocarbanions. As a rationale for the enantiodivergence observed depending on the base used, in the case of less basic hexamethyldisilazides, deprotonation in which a metal cation is pre-complexed with an electrophile was proposed and the proposal was supported by DFT calculations.
Chirality Transfer in Brook Rearrangement-Mediated SE2’ Solvolytic Protonation and Its Use in Estimation of the Propensity for Racemization of the α-Lithiocarbanions of the Substituents
- Chirality transfer from an α-silylalcohol to differently substituted α-carbmoyloxy- and α-siloxyallyl carbanions was investigated using a Brook rearrangement-mediated SE2’ solvolytic protonation in γ-substituted γ-carbamoyloxy- and γ-siloxy-α-silylallyl alcohols. Comparison of the extent of the chirality transfer provides a new method for semi-quantitative evaluation of the propensity for racemization of lithiocarbanions next to a conjugative electron-withdrawing group or an anion-stabilizing heteroatom substituent.
Enantioselective Trapping of an α-Chiral Carbanion of Acyclic Nitrile by a Carbon Electrophile
- An α-chiral nitrile carbanion generated by deprotonation of enantioenriched O-carbamoyl cyanohydrin was trapped in situ with ethyl cyanoformate in Et2O at -114 °C to give the corresponding ester derivative in 92% yield and 90:10 er, providing the first example of trapping of an α-chiral acyclic nitrile carbanion that has been considered to be very configurationally labile.
Steric Course of the Electrophilc Substitution of Lithiocarbanion Generated from (S,E)-1-Phenylbut-2-en-1-yl Diisopropylcarbamate and Influence of Solvent Effects
- The effects of electrophiles and solvents on the stereochemistry of electrophilic substitution of lithiocarbanion generated from (S,E)-1-phenylbut-2-en-1-yl diisopropylcarba-mate were examined using various acids and carbon electrophiles.
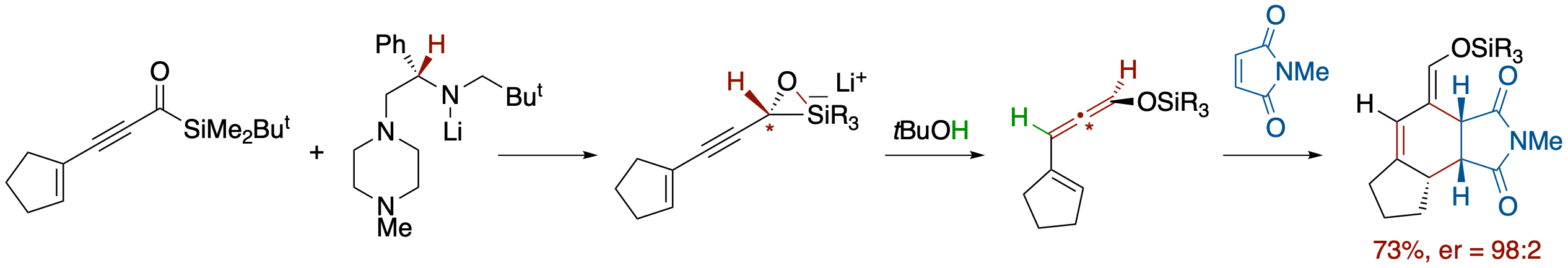
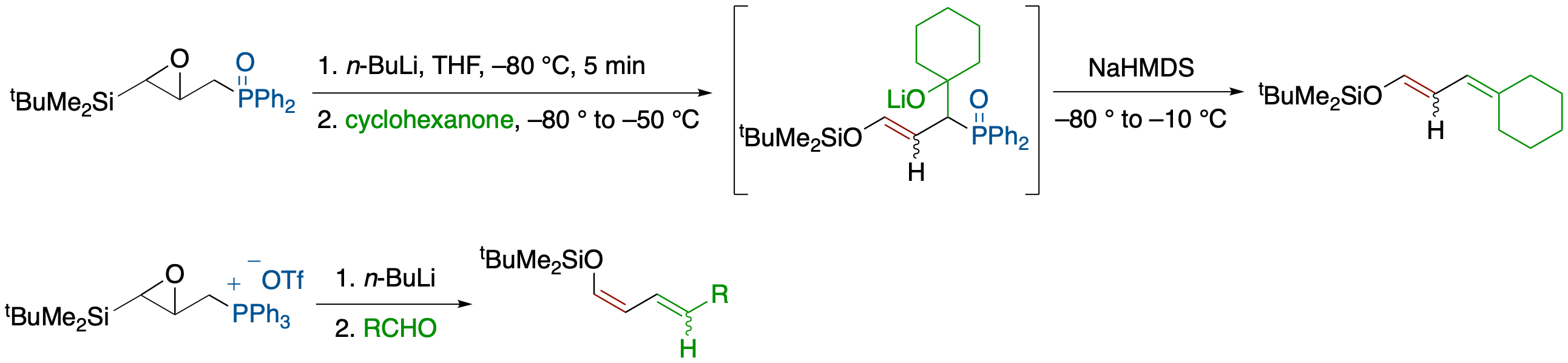
Enantioselective Synthesis of Siloxyallenes from Alkynoylsilanes by Reduction and
Brook Rearrangement and Their Subsequent Trapping in [4 + 2] Cycloaddition
- An enantioselective Meerwein-Ponndorf--Verley-type reduction of alkynoylsilanes by a chiral lithium amide followed by a Brook rearrangement and SE2' electrophilic substitution provides the title compounds in a one-pot process. In the case of enynoylsilanes, the generated vinylallenes undergo in situ [4 + 2] cycloaddition to afford highly functionalized polycyclic compounds with unusual facial selectivity.
Solvent Effects on the Steric Course of [2,3]-Wittig Rearrangement of (S,E)-(3-(allyloxy)prop-1-ene-1,3-diyl)dibenzene and Its Derivatives
- The effect of solvents and additives on configurational stability of chiral carbanions was examined on the basis of extent of chirality transfer in intramolecular trapping in [2,3]-Wittig rearrangement of chiral 1,3-diphenyl-1-propenyloxy-2-propen-1-yl carbanion and its derivatives. The solvent effect was also examined on the Hoffmann test.
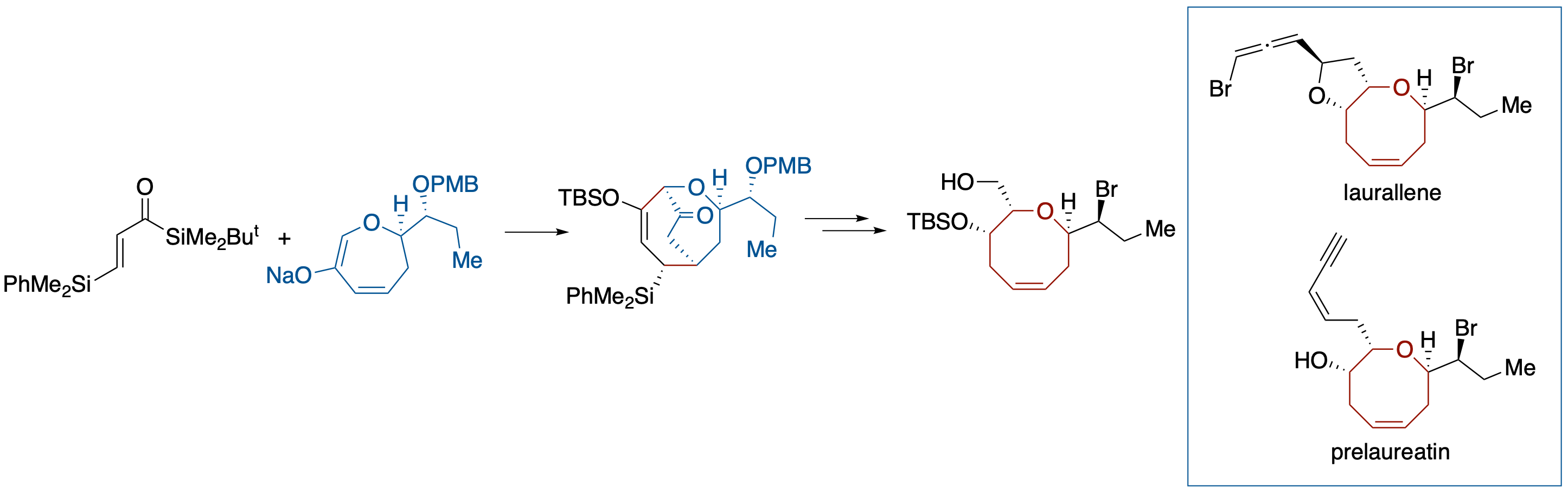
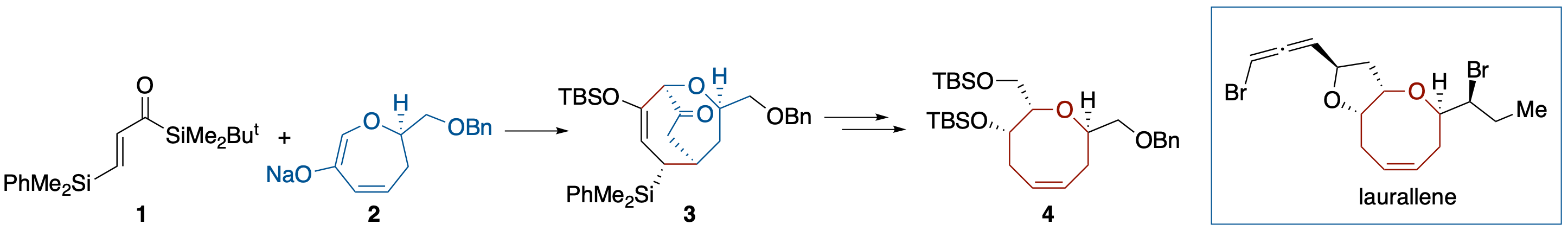
Formal Total Syntheses of (+)-Prelaureatin and (+)-Laurallene by Diastereoselective Brook Rearrangement-Mediated [3 + 4] Annulation
- The formal syntheses of (+)-prelaureatin and (+)-laurallene, halogenated eight-membered-ring ethers, are described. The key step of our strategy relies on diastereoselective construction of a trans-α,α'-disubstituted oxocene structure through a Brook rearrangementmediated [3 + 4] annulation with acryloylsilane and 6-oxa-2-cycloheptenone derivative.
[2,3]-Wittig Rearrangement of Enantiomerically Enriched 3-Substituted 1-Propenyloxy-1-phenyl-2-
propen-1-yl Carbanions: Effect of Heteroatoms and Conjugating Groups on Planarization of an α-Oxy-Benzylcarbanion Through a Double Bond
- The effect of conjugating electron-withdrawing groups and α-anion-stabilizing heteroatom substituents on configurational stability of chiral carbanions through a double bond was examined on the basis of extent of chirality transfer in intramolecular trapping in [2,3]-Wittig rearrangment of chiral 3-substituted 1-propenyloxy-1-phenyl-2-propen-1-yl carbanions.
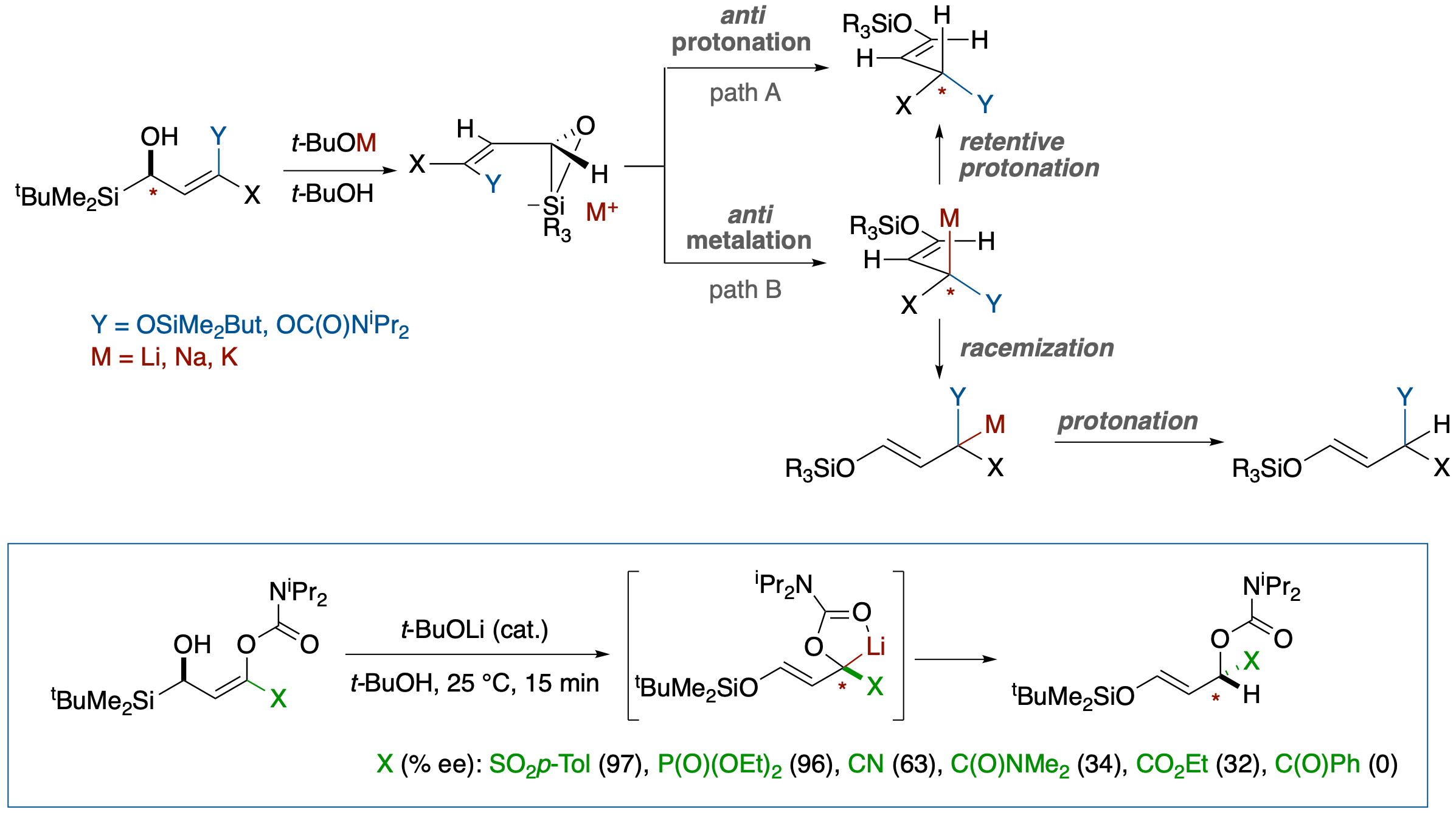
Stereoselective Brook Rearrangement-Mediated SE2' Protonation of α-Hydroxy Allyl Silanes
- Treatment of (R,Z)-3-(tert-butyldimethylsilyl)-1-cyano-3-hydroxyprop-1-enyl carbamate with a catalytic amount of a base afforded (S,E)-3-(tert-butyldimethylsilyloxy)-1-cyanoallyl diisopropylcarbamate, showing that SE2'-type reaction of allylsilicates proceeds in an anti-mode fashion. The overall process is equivalent to trapping of an enantioenriched C-chiral carbanion at a-position of nitrile group in up to 77% ee.
Diastereoselective Brook Rearrangement-Mediated [3 + 4] Annulation: Application to a Formal Synthesis of (+)-Laurallene
- The formal synthesis of (+)-laurallene, a halogenated eight-membered ring ether, was accomplished. The synthesis involves construction of a trans α,α’-disubstituted oxocene structure 3 through a Brook rearrangement-mediated [3 + 4] annulation using acryloylsilane 1 and 6-oxa-2-cycloheptenone 2 and its conversion into 4, which has been transformed into (+)-laurallene by Crimmins and coworkers.
Chirality Transfer from Epoxide to Carbanion: Base-Induced Alkylation of O-Carbamoyl
Cyanohydrins of β-Silyl-α,β-epoxyaldehyde
- Enantioselective C-C bond formation at an α-position of a nitrile group with an external electrophile can be realized, although in modest ee, with the aid of both the concerted process of the epoxysilane rearrangement and a carbamoyl group.
Tandem Epoxysilane Rearrangement/Wittig-Type Reactions Using γ-Phosphinoyl- and γ-
Phosphonio-α,β-epoxysilane
- Reaction of γ-phosphinoyl- and γ-phosphonio-α,β-epoxysilane with a base followed by addition of a ketone or an aldehyde afforded dienol silyl ether derivatives via a tandem process that involves base-induced ring opening of the epoxide, Brook rearrangement, and Wittig-type reaction.
.
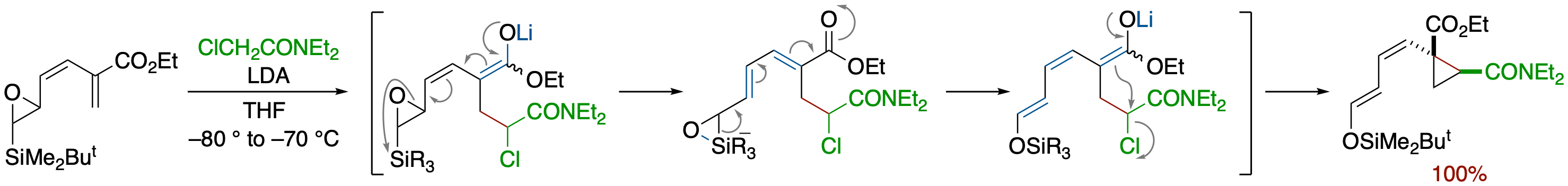
Epoxysilane Rearrangement Induced by a Carbanion Generated by Conjugate Addition of Enolates
of Chloroacetate and α-Chloroacetamides: Formation of Functionalized Cyclopropane Derivatives
- Reaction of an enoate bearing an epoxysilane moiety at the α-position with lithium enolate of 2-chloroacetamide afforded highly functionalized cyclopropane derivatives via a tandem process that involves Michael addition, ring opening of the epoxide, Brook rearrangement, and intramolecular alkylation.
Asymmetric [2,3]-Wittig Rearrangement Induced by a Chiral Carbanion Whose Chirality Was Transferred from an Epoxide
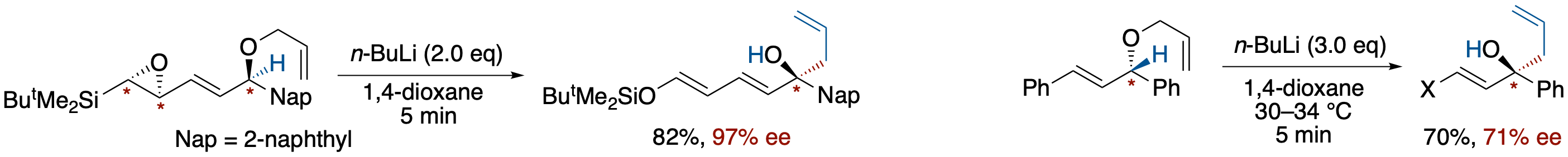
- The enantioselective [2,3]-Wittig rearrangement of 1-allyloxy-1-(naphthalen-2-yl)-4-siloxy-2,4-pentadienyl anion, derived from optically enriched 4,5-epoxy-1-(naphthalen-2-yl)-5-silyl-2-pentenyl allyl ether via a base-induced ring opening of the epoxide followed by Brook rearrangement, has been studied. The chirality of the epoxide was transferred to the alcohols in up to 97% ee, depending on the solvent used. The best result was obtained in 1,4-dioxane at a temperature above room temperature.
γ-p-Toluenesulfonyl-α,β-epoxysilane: A New and Practical Acrolein β-Anion Equivalent
- Reaction of γ-p-toluenesulfonyl-α,β-epoxysilane with alkyl halides and aldehydes followed by treatment with n-Bu4NF affords α,β-unsaturated aldehydes via a Brook rearrangement-mediated tandem process under extremely mild conditions.
Stereoselective Construction of Eight-Membered Oxygen Heterocycles by Brook Rearrangement-
Mediated [3 + 4] Annulation
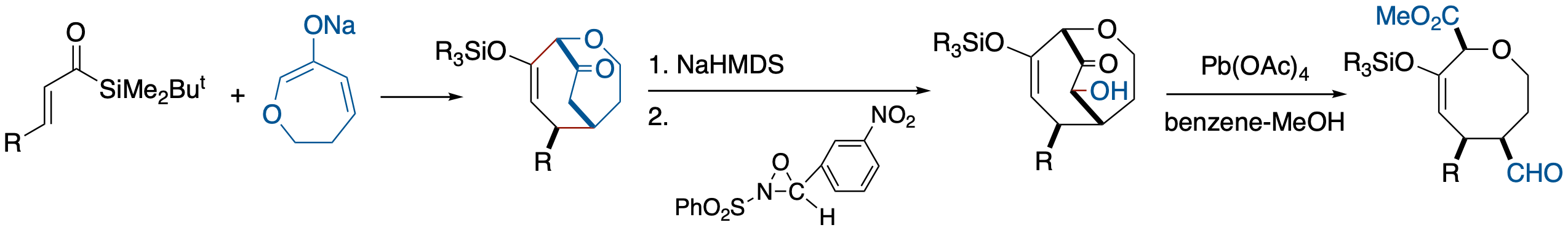
- A newly developed strategy for eight-membered oxgen heterocycles via [3 + 4] annulation that involves the combination of β-substituted acryloylsilanes and enolates of 6-oxacyclohept-2-en-1-one is described. A unique feature of this annulative approach is its capacity to generate eight-membered ring systems containing useful functionalities for further synthetic elaboration from readily available three- and four-carbon components.
Tandem Base-Promoted Ring-Opening/Brook Rearrangement/Allylic Alkylation of O-Silyl
Cyanohydrins of β-Silyl-α,β-epoxyaldehyde: Scope and Mechanism
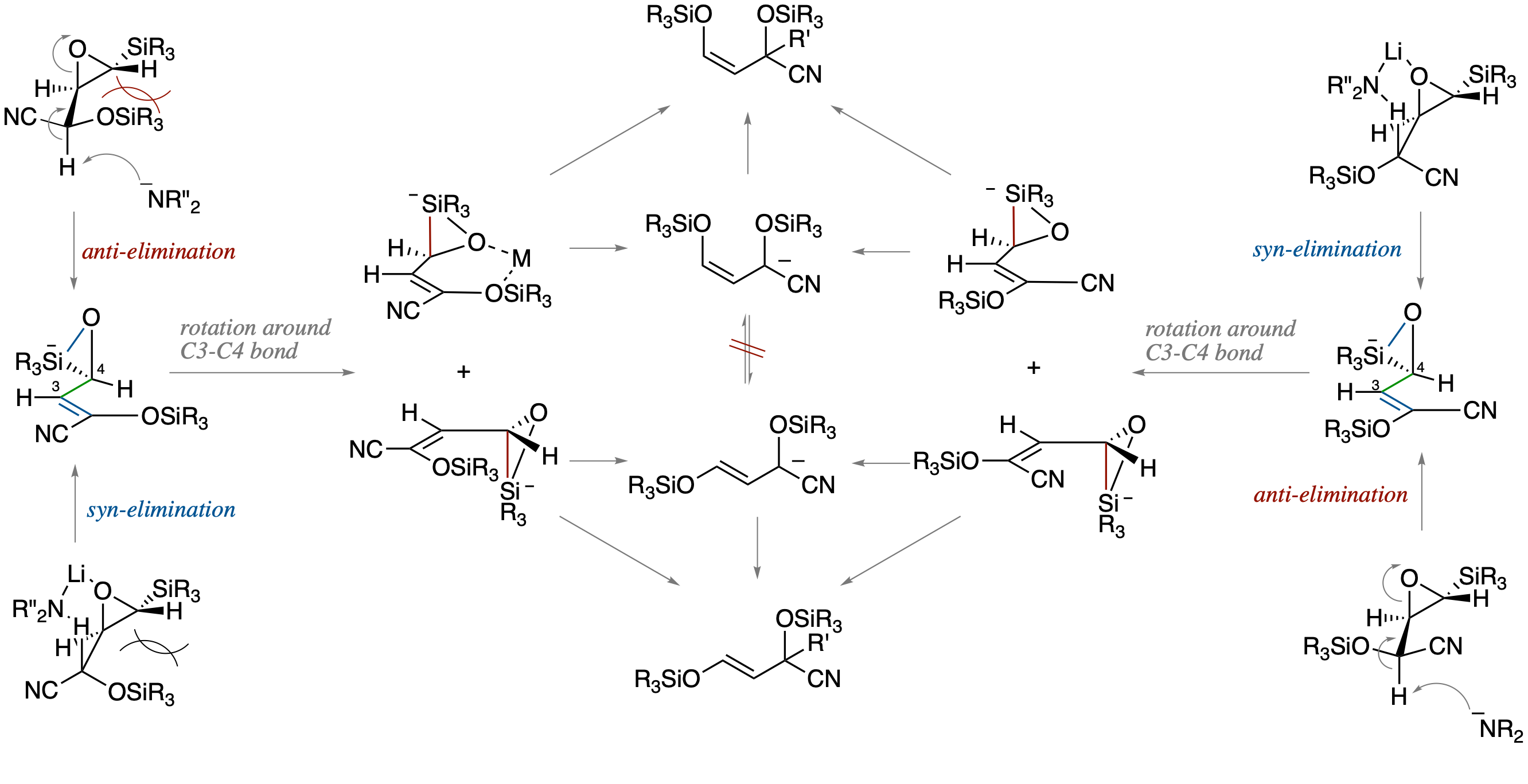
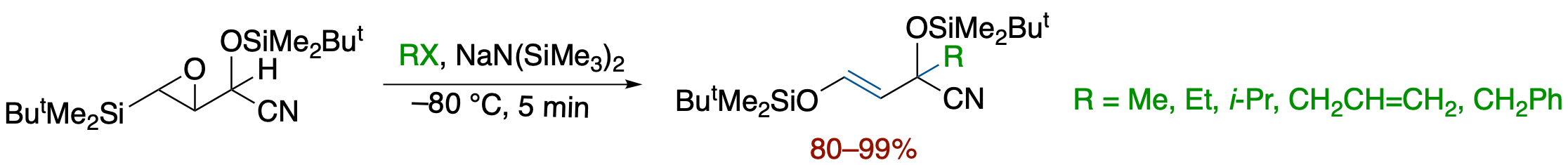
- Metalated O-silyl cyanohydrins of β-Silyl-α,β-epoxyaldehyde have been found to serve as functionalized homoenolate equivalents by a tandem sequence involving base-promoted ring opening of the epoxide, Brook rearrangement, and alkylation of the resutling allylic anion. On the basis of mechanistic studies involving competitive experiments using the diastereomeric cyanohydrins, we propose a reaction pathway involving a silicate intermediates formed by a concerted process via an anti-opening of the epoxide followed by the formation of an O-Si bond.
Tandem Base-Promoted Ring-Opening/Brook Rearrangement/Allylic Alkylation of O-Silyl
Cyanohydrins of β-Silyl-α,β-epoxyaldehyde
- Metalated O-silyl cyanohydrins of β-Silyl-α,β-epoxyaldehyde have been found to serve as functionalized homoenolate equivalents by a tandem sequence involving a base-promoted ring opening of the epoxide, Brook rearrangement, and alkylation of the resulting allylic anion.