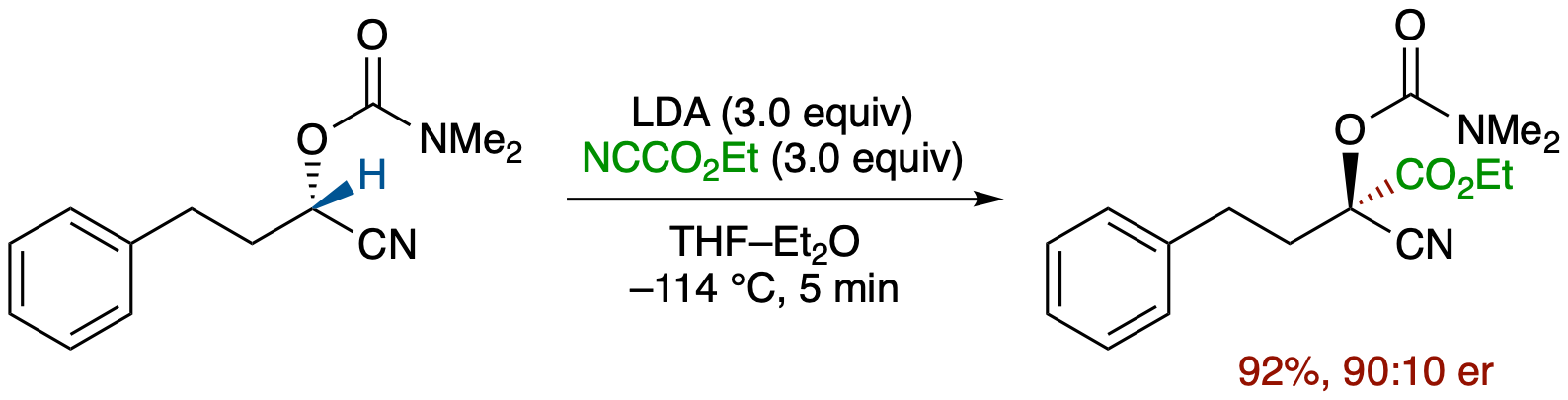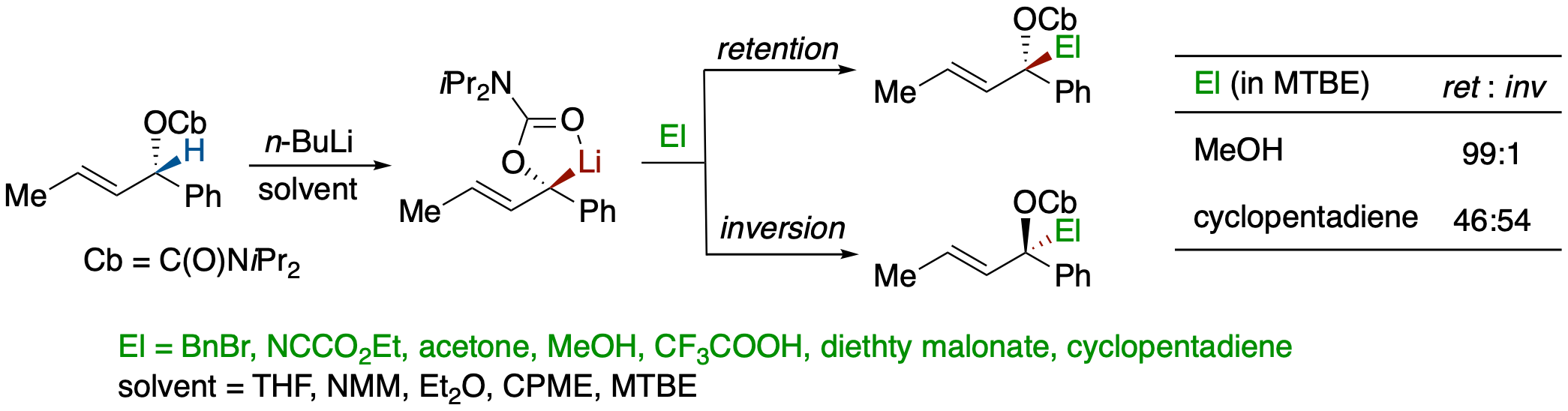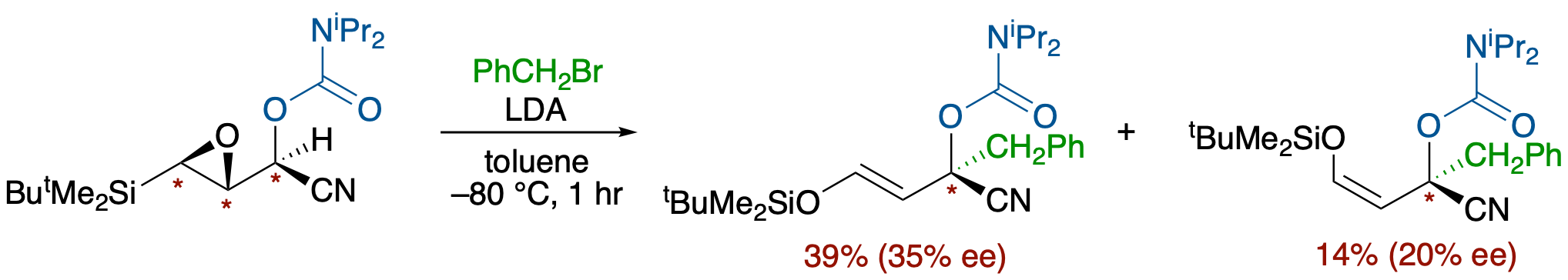Research
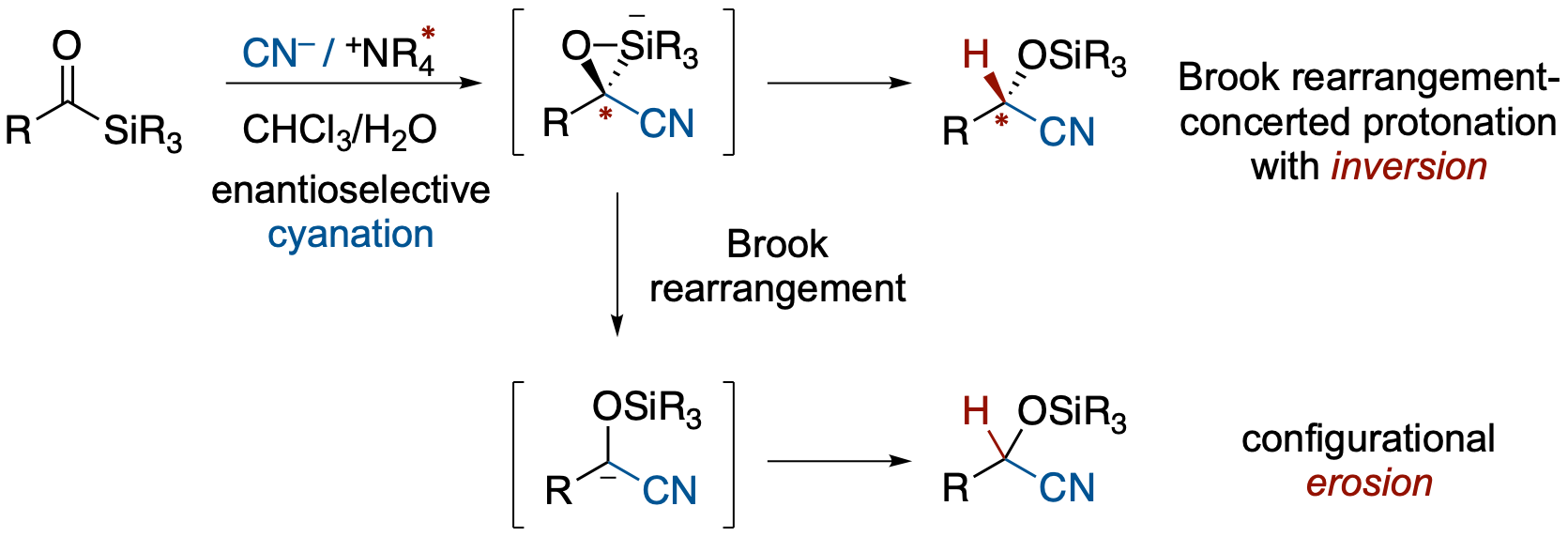
アシルシランのエナンチオ選択的シアノ化–Brook転位–プロトン化反応とBrook転位の立体化学
- アシルシランに対してキラルな相間移動触媒存在下,二相系溶媒中シアニドイオンを反応させると,シアノ化,Brook転位,プロトン化が連続的に進行し,O-シリルシアノヒドリンが最大92:8 erで得られた.別途合成したα-シリルシアノヒドリンを用いた検討により,形式的なα-ニトリルカルバニオン等価体を生成するBrook転位はinversionで進行することが明らかになった.また,対カチオン,同位体効果の検討,クロスオーバー実験の結果などから,全体のエナンチオ選択性は主に初期のシアノ化段階で導入されるが,シリケートからの協奏的なプロトン化と,非局在化したα-ニトリルアニオンを経由する段階的過程との競合によって最終的な選択性が決定されることが示唆された.
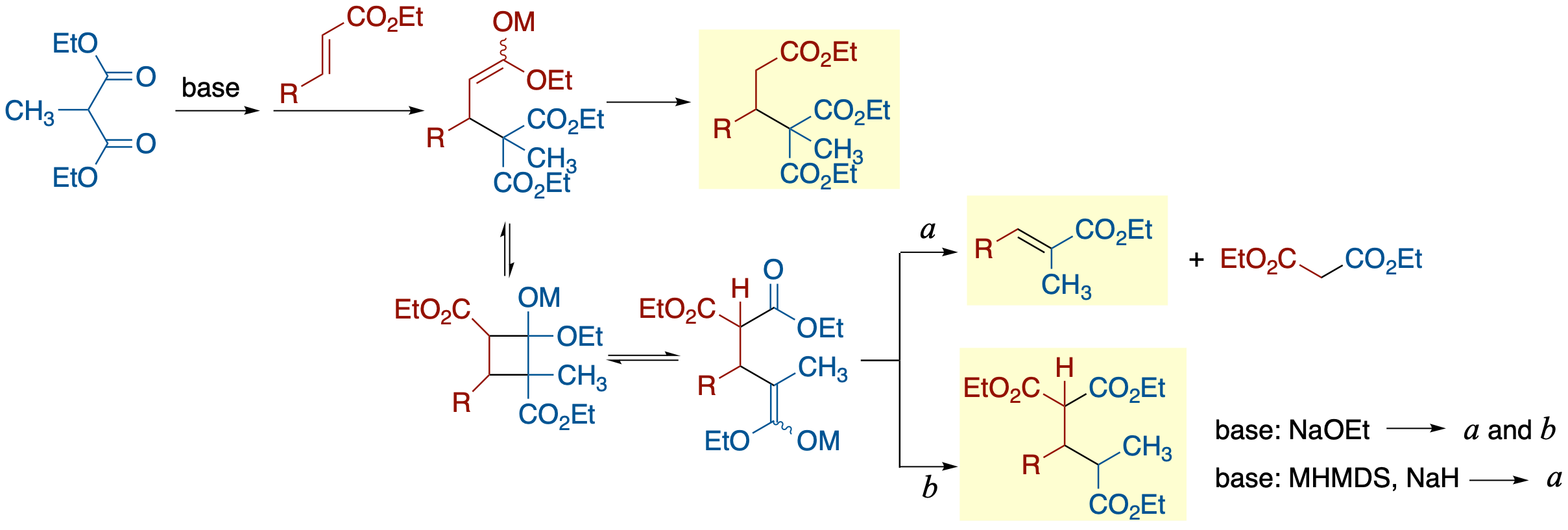
モノアルキル置換マロネートのマイケル反応における生成物の条件依存性
- abnormal Michael反応は,Michael反応において求核種としてモノアルキル置換マロネートを用いると,求核種のエステル基がマイケル受容体のα-炭素に転位するという反応であり,1900年にThorpeおよびMichaelによって報告された.本反応について,分子内反応,分子間反応両方を検討した結果,以下の事実が明らかになった.
- 系内にプロトン源が無い場合(塩基:NaHMDS, NaH, 溶媒:THF, Et2O),abnormal Michael成績体は得られず,エステル基の転位の後にマロネートアニオンが脱離した化合物が主成績体となる.
- 系内にプロトン源が存在する場合(塩基:NaOEt),分子間反応の場合のみ,abnormal Michael成績体が得られる.ただし,脱離体が主成績体である.
- これらのことから,マロネートの速度論的酸性度は熱力学的酸性度から予測されるよりも低いこと,abnormal Michael成績体はアルコールが介在したプロトン移動によって生成していることが示唆された.
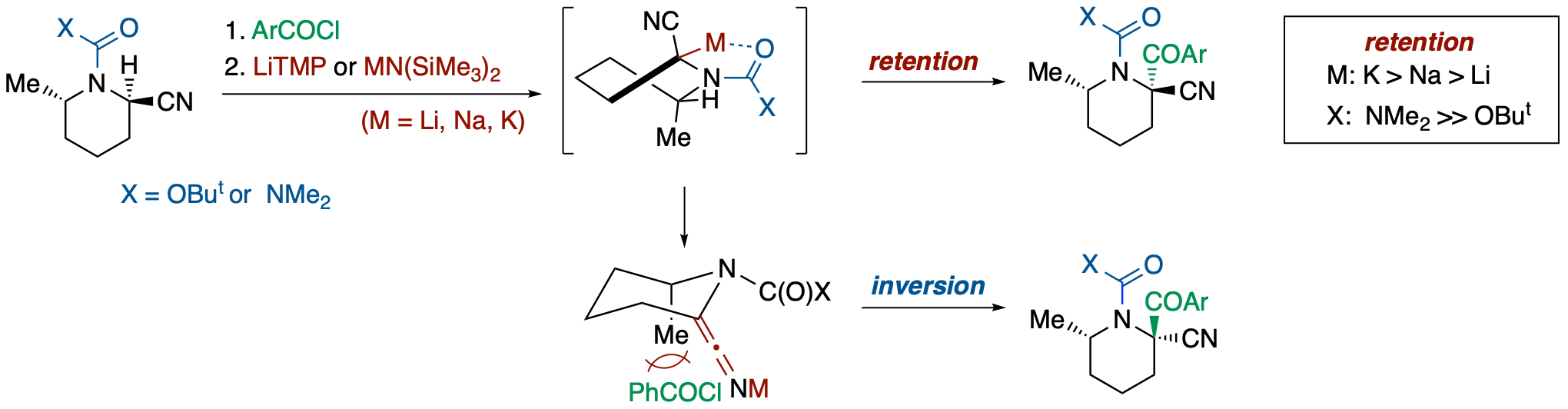
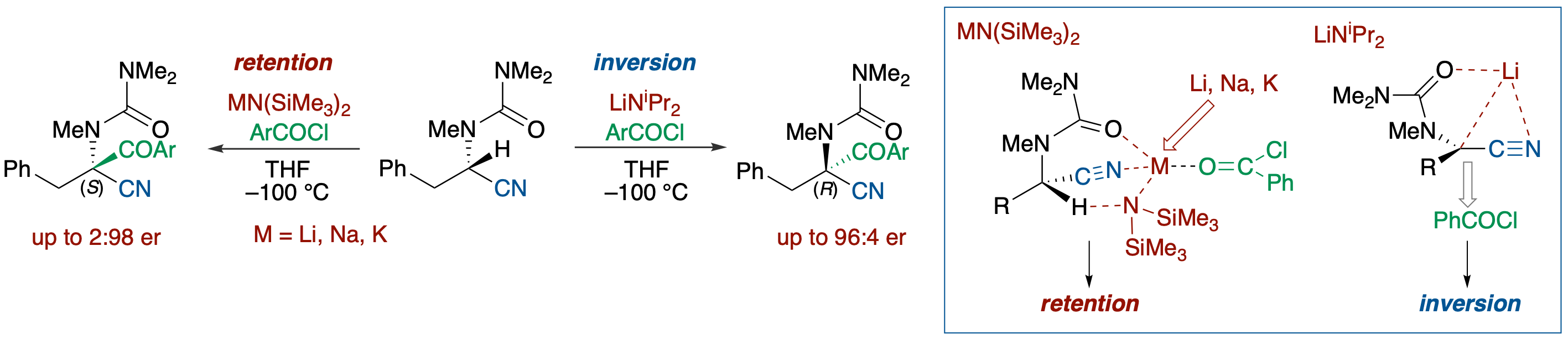
双極子安定化置換基を有するα-アミノおよびα-オキシニトリルの脱プロトン化/置換反応の立体過程
- エナンチオ過剰のカルバモイル基もしくはメトキシカルボニル基を有するα-アミノ,α-オキシニトリルを塩化ベンゾイルの存在下アミド塩基で脱プロトン化し,得られたベンゾイル化体のエナンチオマー比を比較した.また,脱プロトン化の速度の比較,塩化ベンゾイルのベンゼン環上の置換基が及ぼす影響の検討,計算科学により求めたアニオン種の構造の比較などを行った結果,双極子安定化置換基や塩基の違いが,α-ニトリルカルバニオンの立体化学的安定性および求電子剤に対する反応性だけではなく,求電子剤との反応における立体過程(立体保持/立体反転)に影響を及ぼしていることが明らかになった.
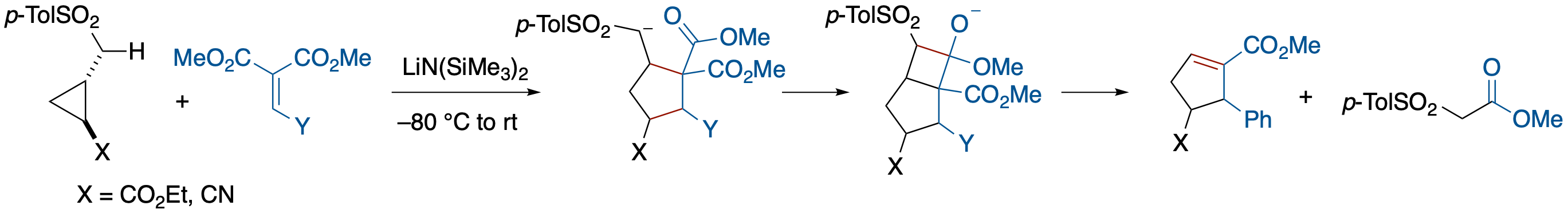
カルバニオンをドナーとするD-Aシクロプロパンの[3+2]アニュレーション
- p-トシルメチルシクロプロパン誘導体を,イリデンマロネート存在下,LiN(SiMe3)2で処理すると,シクロペンテン誘導体が得られる.本反応では,分子間・分子内マイケル反応の後に,形式的なcycloreversionが起こってトシルアセテートが脱離したと考えられる.
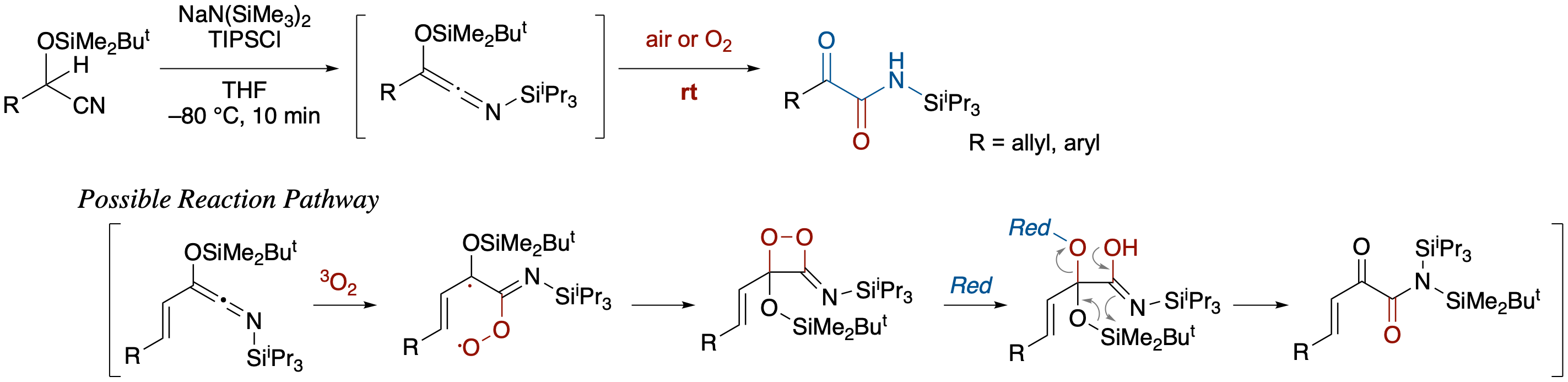
Siloxy-N-Silylketenimine のα-ケトアミドへの空気酸化
- O-シリルシアノヒドリンに対し NaN(SiMe3)2/i-Pr3SiCl (TIPSCl) を反応させることにより発生させたシロキシ-N-シリルケテンイミンを,空気もしくは酸素で処理するとα-ケトアミドが生成する.極めて緩和な条件下,酸化が進行する機構として,シロキシ-N-シリルケテンイミンと三重項酸素との反応後,三重項ー一重項の項間交差で生成する3-イミノ-1,2-ジオキセタン中間体を経る機構が提出された.
N-Boc- および N-carbamoyl-2-cyano-6-methylpiperidines の脱プロトン化/アシル化反応の立体過程
- α-ニトリルプロトンの高い酸性度に着目して,N-Boc- および N-carbamoyl-2-cyano-6-methylpiperidines 対しアシル化剤の存在下塩基を加えたところ,配位性置換基,対カチオン,求電子剤の反応性に依存して SE2 反応の立体過程が変化することが明らかになった.すなわち,
- SE2 反応は基本的に立体保持で進行する.
- 窒素上の配位性置換基(Boc, Cb)の配位能が大きいほど,また対カチオン(Li, Na, K)の脱離能(イオン性)が高いほど,立体保持成績体の割合が増大する.
- 反応性が高い求電子剤(o-ClC6H4COCl > C6H5COCl > p-MeOC6H4COCl)ほど立体保持で進行する割合が高くなる.
キラルリチウムアミドによるイネノイルシランの不斉還元を利用するアレニルエノールシリルエーテルの合成とそのDiels-Alder反応
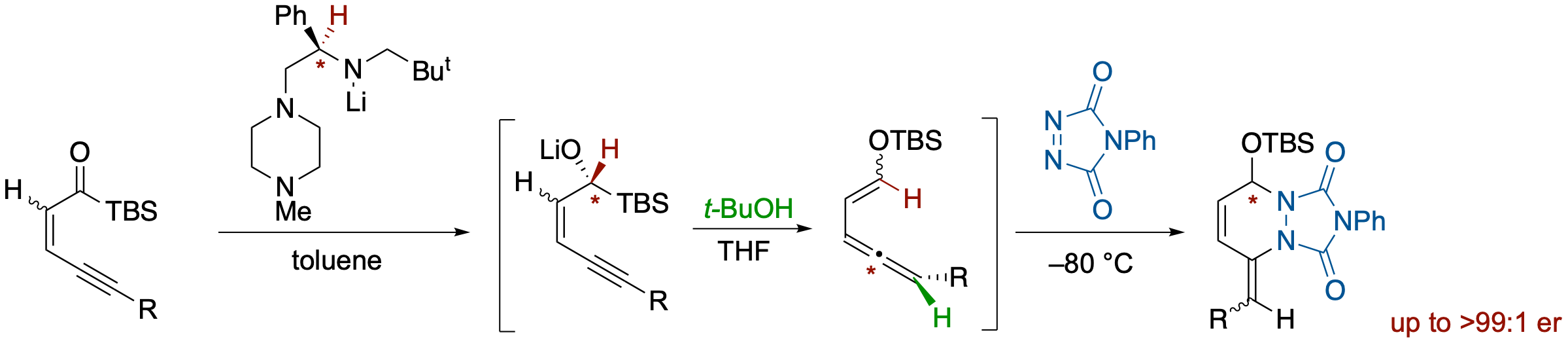
- イネノイルシランをキラルリチウムアミドで処理するとMeerwin-Ponndorf-Varleyタイプの還元がおこり,さらにt-BuOH/THFを加えることでBrook転位/1,3-エンインを経由するanti選択的なプロトン化が進行し,アレニルエノールシリルエーテルが高いエナンチオマー比で得られた.エノールシリルエーテルのE/Z-ジオメトリーはイネノイルシランのE/Z-ジオメトリーによってコントロールされ,E体からはZ体の,Z体からはE体のエノールシリルエーテルを有する成績体が得られた.得られたシロキシビニルアレンはDiels-Alder反応の基質となり,PTADなどと反応させることでワンポットで環状化合物に導くことが可能である.
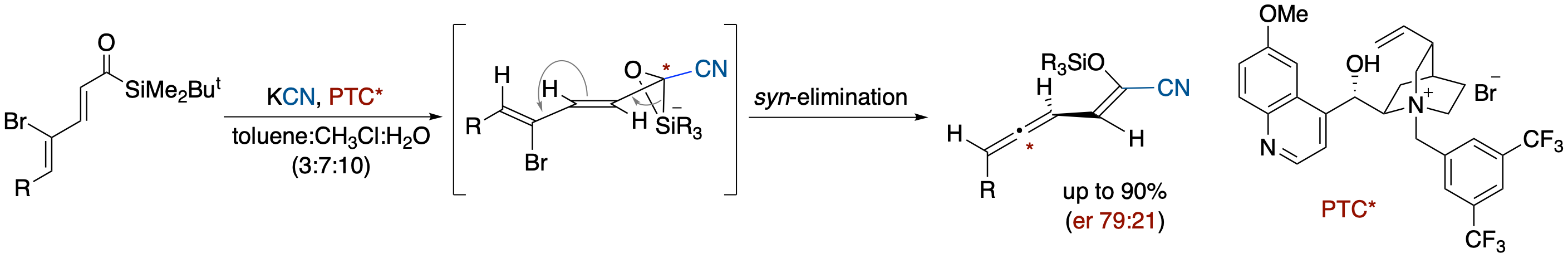
γ-ブロモ-α,β,γ,δ-不飽和アシルシランに対するシアニドイオンのエナンチオ選択的付加を利用する2-シアノ-2-シロキシビニルアレンの合成
- γ-ブロモ-α,β,γ,δ-不飽和アシルシランに,相関移動触媒としてシンコナアルカロイド由来の四級アンモニウム塩存在下,二層系溶媒中KCNを反応させると,2-シアノ-2-シロキシビニルアレンが最大エナンチオマー比79:21で得られた.また,このようなBrook転位を経由する1,4-脱離反応は,シリケート中間体において切断されるC-Si結合とC-Br結合が同じ向きを向いた配座から進行するsyn脱離でおこっていることが明らかになった.
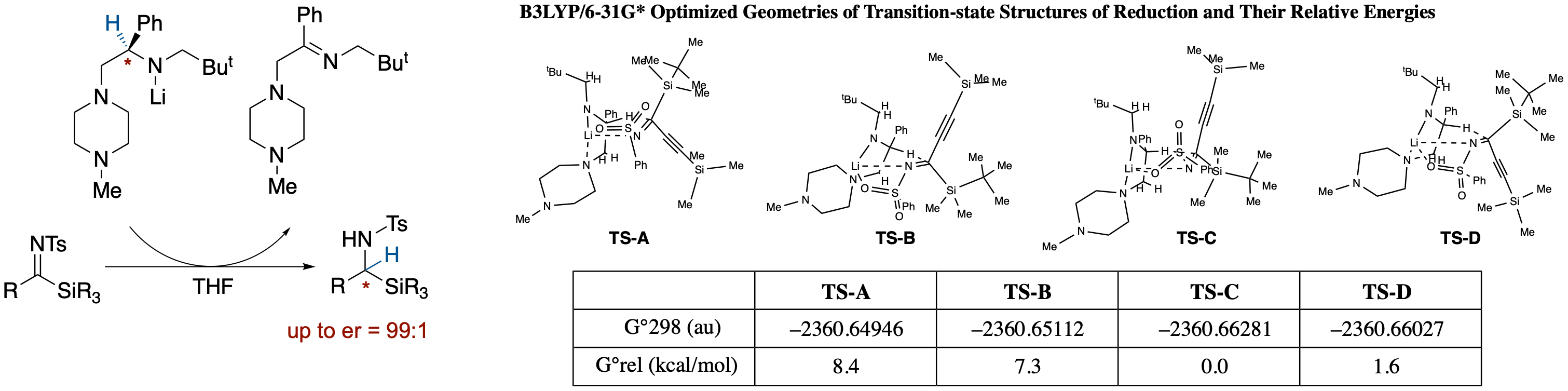
α-シリルイミンの不斉還元によるα-シリルアミンの合成
- N-トシルシリルイミン1をキラルリチウムアミド2で処理すると Meerwin-Ponndorf-Varleyタイプの還元がおこり,α-シリルアミンが高いエナンチオマー比で得られた.成績体の立体化学が,イス型の六員環遷移状態構造では説明できないため,遷移状態のDFT計算(モデル化合物)を行ったところイス型から大きくずれており,1'-メチレン基とイミン炭素上の置換基の立体反発がエナンチオ選択性に大きな影響を及ぼしていることが明らかになった.
キラルα-ニトリルカルバニオンのエナンチオダイバージェントな脱プロトン化-アシル化
- α-アミノ酸から容易に得られるエナンチオ過剰α-ウレイドニトリルをアロイルクロリドの存在下アミド系塩基を作用させると,極めて立体化学的に不安定なα-ニトリルカルバニオンを経由するにもかかわらず,脱プロトン化-アシル化が高エナンチオ選択的に進行する.特に注目すべき点は,LDAでは立体反転で,NaHMDS, LiHMDS, KHMDSでは立体保持で進行することで,この結果を説明するために,より塩基性が小さなヘキサメチルジシラジドの場合,アロイルクロリドが塩基の対カチオンとコンプレックスを形成した状態で脱プロトン化が起こるのに対して,より塩基性が大きなジイソプロピルアミドでは,脱プロトン化にアロイルクロリドは関与せずにリチオカルバニオンが生成し,より立体反発の少ないリチウムの逆側からアロイルクロリドが反応するという仮説が提出した.この仮説はDFT計算により支持された.
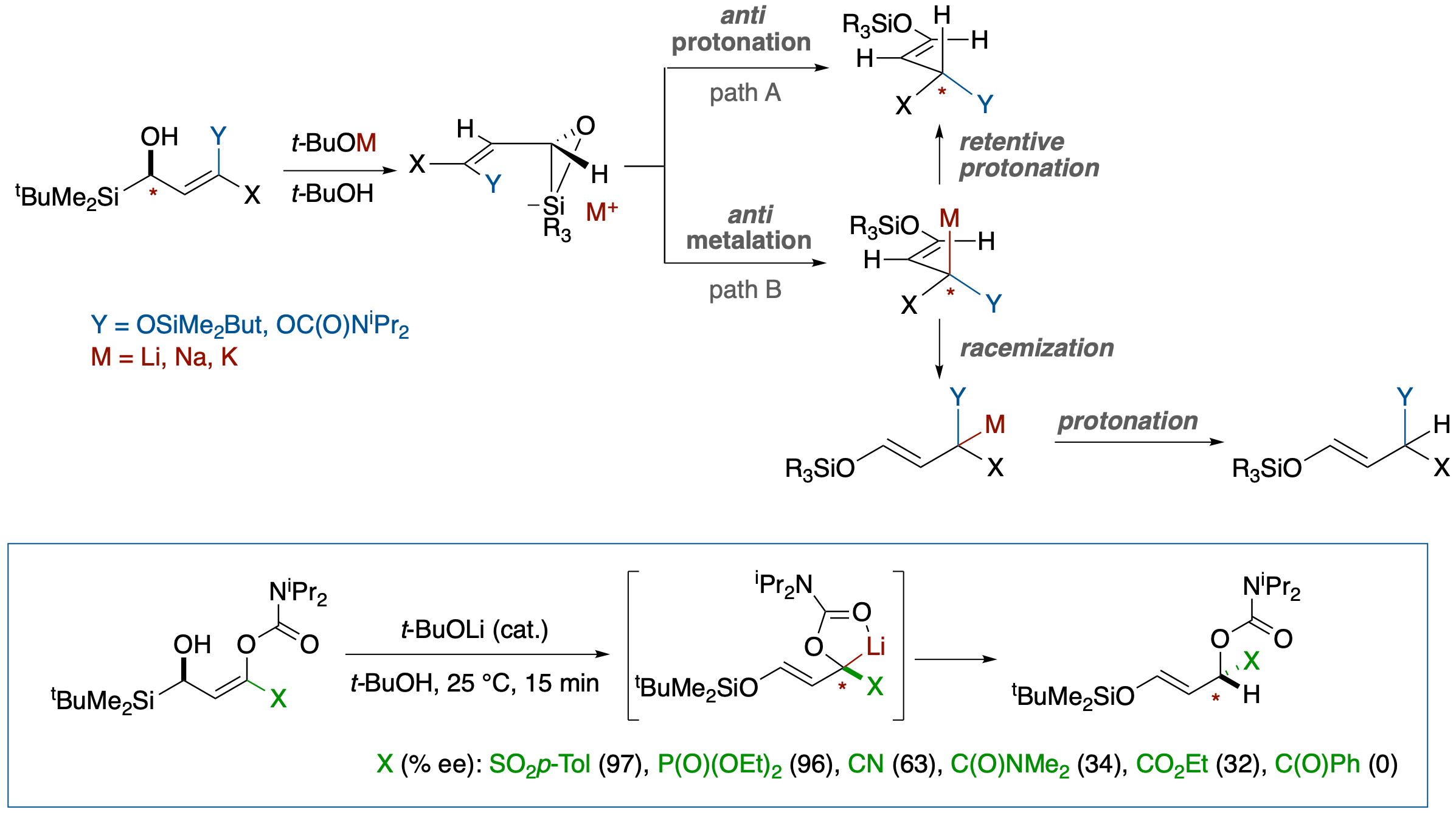
Brook転位を経るSE2'型加溶媒プロトン化における不斉転写の反応機構とそれを利用する電子求引性置換基に隣接するキラルカルバニオンのラセミ化傾向の半定量的評価
- ヒドロキシアリルシランのBrook転位を経るSE2’型のプロトン化反応における不斉転写の機構に関し,(1) 置換基(X, Y)効果, (2) プロトン源についての同位体効果(t-BuOH, t-BuOD),(3) 対カチオンの効果(Li, Na, K)(4) 溶媒効果,を検討し,次のような結論を得た.
置換基Yのカチオンに対するキレーション能の大きさに依存し,シリケート中間体の協奏的プロトン化を経由するPath Aと,シリケート中間体から協奏的メタル化を経てメタロカルバニオン中間体が生成した後プロトン化されるPath B,の二つの経路で進行する.
キレーション能の小さなシロキシ基の場合Path Aを,キレーション能の大きなカルバモイルオキシ基の場合はキラルメタロカルバニオンを中間体とするPath Bを経由する.
カルバモイル誘導体の場合,キラルなリチオカルバニオンを経由することに基づき,電子求引性置換基 X が隣接位のキラルカルバニオンのラセミ化に及ぼす影響を半定量的に評価する方法を開発した.
その結果,p-TolSO2, (EtO)2P(O), PhMe2Si, CN, Me2NC(O), EtO2C, PhC(O)の順に,ラセミ化しやすいということが,明らかになった.
脱プロトン化により発生させたキラルα-ニトリルカルバニオンの炭素求電子剤による捕捉
- これまで立体化学的に極めて不安定なため発生が不可能と考えられてきた鎖状ニトリルのα-キラルカルバニオンの発生および炭素求電子剤による捕捉に成功した.すなわち,O-カルバモイルシアノヒドリン誘導体を,Et2O-THF中シアノギ酸エチル存在下-114℃においてLDAで処理したところ,er = 90:10 でエステル化体を立体反転で与えた.
(S,E)-1-Phenylbut-2-en-1-yl Diisopropylcarbamateから発生させたリチオカルバニオンの求電子置換反応の立体過程とそれに対する溶媒効果
- 立体化学的に不安定と予想される,(S,E)-1-phenylbut-2-en-1-yl diisopropylcarbamate から発生させたリチオカルバニオンの求電子置換反応の立体化学を,種々のエーテル系溶媒中,炭素求電子剤およびプロトン化剤を用いて検討した.その結果,
- 上記キラルカルバニオンはエーテル性溶媒中-50℃,5分という条件ではラセミ化しない.
- 炭素求電子剤は inversion で,プロトン化剤は retention で反応する割合が高いが,求電子剤および溶媒のリチウムイオンに対するキレーション能の違いによりその割合が変化する.
- ということが明らかになった.
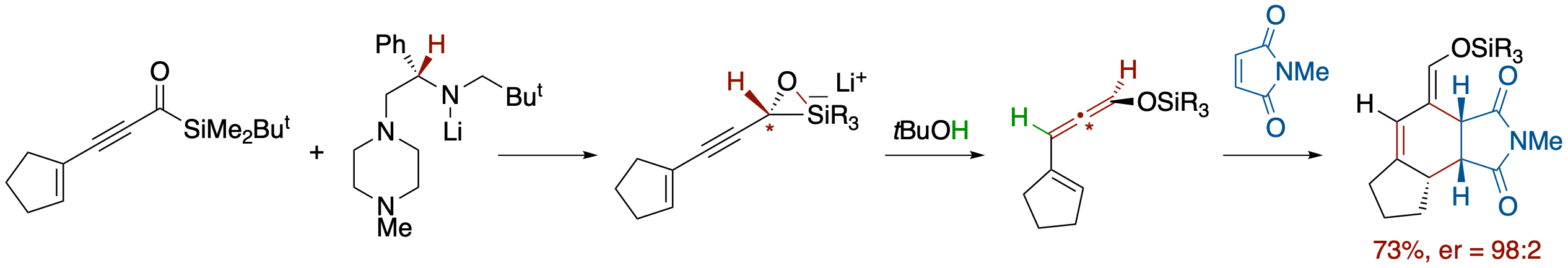
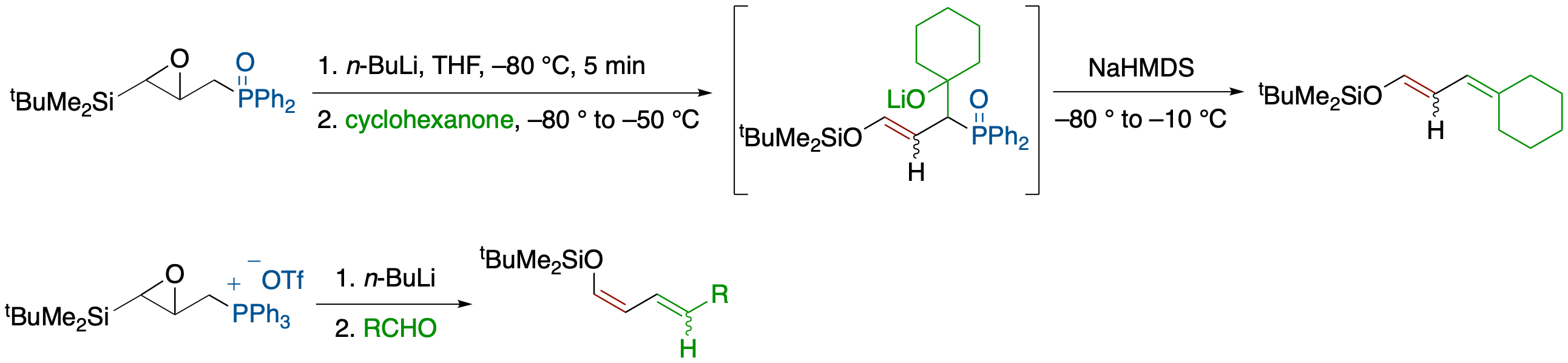
アルキノイルシランの不斉還元/Brook 転位を経由する光学活性シロキシアレンの合成及び[4 + 2]環化付加反応によるその捕捉
- アルキノイルシランをキラルリチウムアミドで処理すると,Meerwin-Ponndorf-Varleyタイプの還元がおこった後,Brook転位/SE2’型の求電子置換反応が進行し,光学活性シロキシアレンがワンポットで得られることを見いだした.さらに,エニノイルシランを原料として用いた場合,得られたビニルアレンを系内で[4 + 2] 環化付加反応によって捕捉することで,多官能性の縮環化合物を高い選択性で得ることに成功した.また,環化反応における面選択性は立体的要因から推測されるものと逆であるという興味深い知見が得られた.
(S,E)-(3-(allyloxy)prop-1-ene-1,3-diyl)dibenzeneおよびその誘導体の [2,3]-Wittig 転位における溶媒効果
- キラルな1,3-diphenyl-1-propenyloxy-2-propen-1-ylカルバニオンの[2,3]-Wittig転位における不斉誘起の程度に基づきキラルカルバニオンの立体化学的安定性に対する溶媒およびadditiveの効果を検討した.また,観察された溶媒効果の一般性を検証する目的でを,分子間反応を用いるHoffmannテストに対する効果も併せて検討した
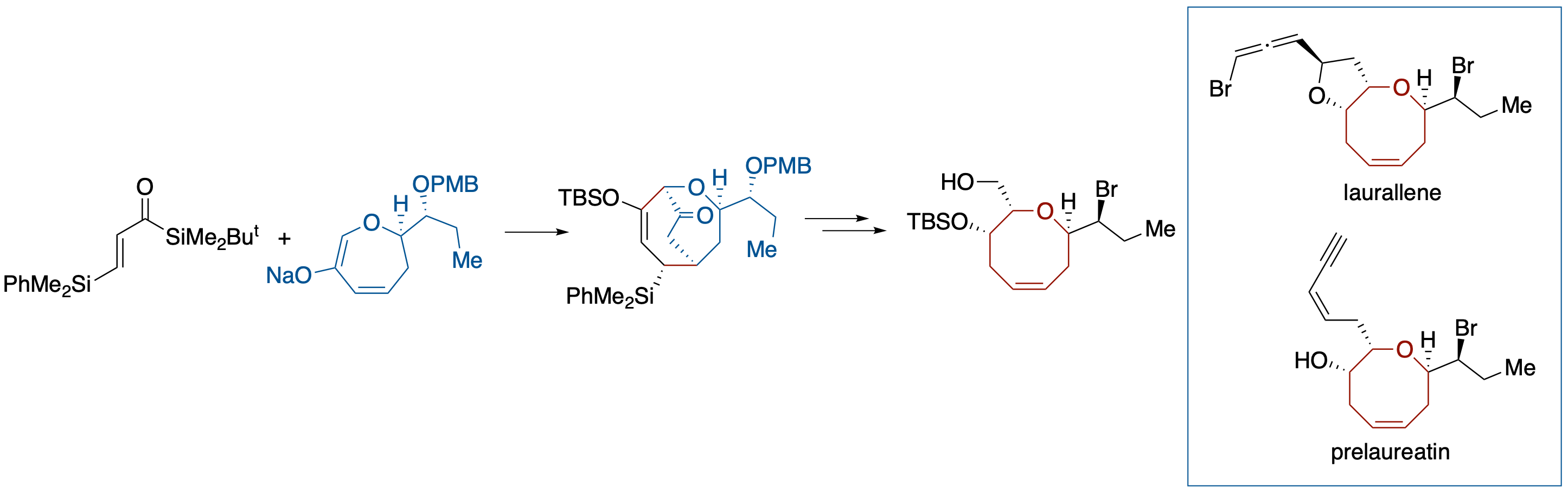
ジアステレオ選択的 Brook 転位介在 [3 + 4] アニュレーションを利用する (+)-Prelaureatin and (+)-Laurallene の形式全合成
- アクリロイルシランと 6-オキサ-2-シクロヘプテノン誘導体の Na エノレートとの Brook転位介在 [3 + 4] アニュレーションによりジアステレオ選択的に生成したビシクロ[3.3.2]誘導体に対し官能基変換を行うことにより,(+)-Prelaureatin および (+)-Laurallene に導くことに成功した.
[2,3]-Wittig転位を利用するキラルカルバニオンの立体化学的安定性の評価
- 光学活性な3位置換1-propenyloxy-1-phenyl-2-propene-1-yl carbanion の [2,3]-Wittig転位における不斉転写の程度に基づき,共役性電子求引性置換基およびα-アニオンを安定化するヘテロ原子置換基 X の,二重結合を通したカルバニオンの立体化学的安定性に対する効果を評価できることを見出した.
Brook転位を経由するα-ヒドロキシアリルシランの立体選択的SE2'型プロトン化
- 光学活性な3-(tert-butyldimethylsilyl)-1-cyano-3-hydroxyprop-1-enyl carbamateを触媒量の塩基で処理すると,アリルシリケートの形成/SE2'型プロトン化がおこり,ニトリルのα位に最大77% eeで不斉が転写されることを見いだした.また,生成物の立体化学から,アリルシリケートのSE2'型プロトン化はantiモードで進行することが判明した.
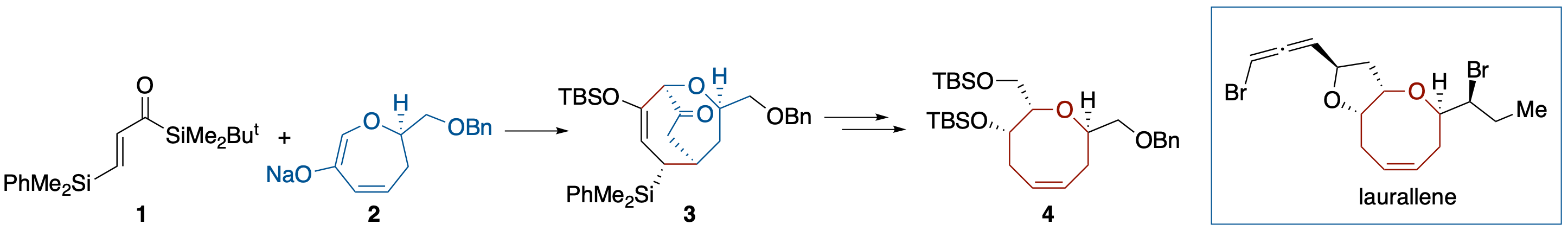
ジアステレオ選択的 Brook 転位介在 [3 + 4] アニュレーション:(+)-Laurallene の形式全合成
- アクリロイルシラン 1 と 6-オキサ-2-シクロヘプテノンのエノレート 2 との Brook転位介在 [3 + 4] アニュレーションによりジアステレオ選択的に生成したビシクロ[3.3.2]誘導体 3 に対し官能基変換を行うことにより,4 に導くことに成功した.4 は Crimmins らによって含臭素八員環エーテル構造を有する (+)-Laurallene に変換されている.
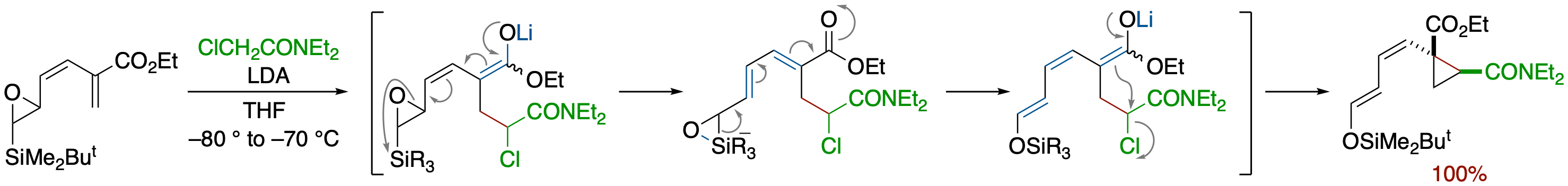
エポキシドからカルバニオンへの不斉転写:β-シリル-α,β-エポキシアルデヒドのO-カルバモイルシアノヒドリンのアルキル化反応
- エポキシシラン転位の協奏的過程とカルバモイル基のキレーション能を利用することにより,エポキシドのキラリティをシアノ基のα位に転写することに成功した.
γ-ホスフィノイルおよびγ-ホスホニオ-α,β-エポキシシランを用いるタンデムエポキシシラン転位/Wittig 型反応
- γ位にホスフィノイルおよびホスホニオ基を有するα,β-エポキシシランを塩基で処理した後ケトンあるいはアルデヒドを加えると,エポキシシラン転位に引き続き Wittig タイプの反応が連続的に進行し,ジエノールシリルエーテルを与える.
α-クロロアセトアミドのエノレートの共役付加によって生成したカルバニオンによって誘起されるエポキシシラン転位: 多官能性シクロプロパン誘導体の生成
- α位にエポキシシラン部位を有するアクリレートに対し,α-クロロアセトアミドのエノレートを反応させると,共役付加によって生成したエノレートアニオンに誘起されてエポキシシラン転位がおこった後,生じたエノレートアニオンによる分子内求核置換反応が進行して,多官能性のシクロプロパン誘導体が高収率で生成する.
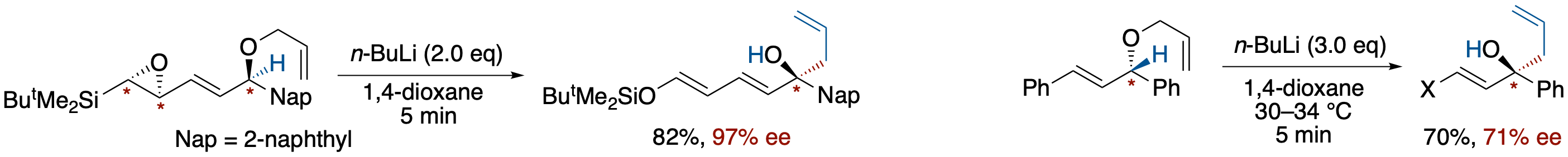
エポキシドからカルバニオンへの不斉転写を鍵とする不斉 [2,3]-Wittig 転位
- 1-allyloxy-1-(naphthalen-2-yl)-4-siloxy-2,4-pentadiene を n-BuLi で処理すると,(1) アニオンによるエポキシドの開環, (2) シリケート中間体の生成, (3) Brook 転位, (4) [2,3]-Wittig 転位を含むタンデム過程が進行し,高収率でアルコール体を与える.ホモキラルなエポキシドを用いると,THF 中ではラセミ化するのに対し,1,4-dioxane 中では最高97% の不斉収率が観察された.また,アリル基とアリール基に挟まれた極めてラセミ化しやすいカルバニオンでも,1.4-dioxane 中では [2,3]-Wittig 転位によって十分捕捉され得る程度の寿命を持つことが明らかになった.
新規アクロレインβ-アニオン等価体:γ-p-Toluenesulfonyl-α,β-epoxysilane
- γ-p-トルエンスルフォニル-α,β-エポキシシランがアクロレインβ-アニオン等価体として機能することを見いだした.すなわち,NaN(SiMe3)2 で処理し,ハロゲン化アルキル,もしくはアルデヒドと反応させた後にTBAFを加えると,-80 ℃から-60 ℃という極めて穏和な条件下,高収率でα,β-不飽和アルデヒドが得られる.
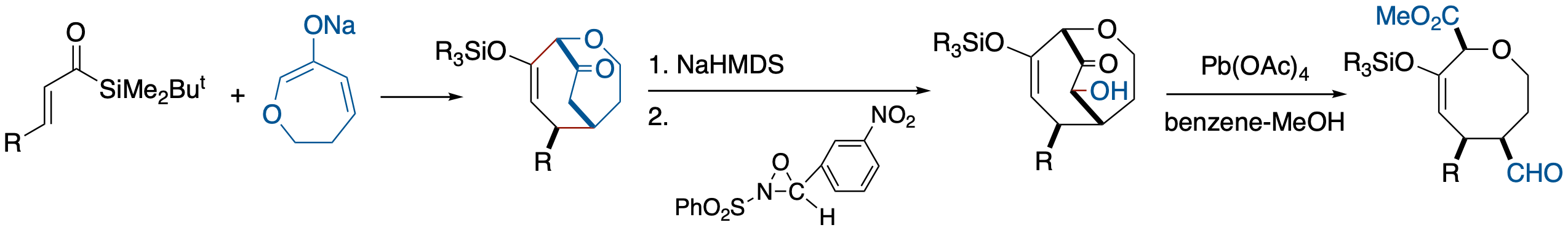
[3 + 4] アニュレーションを利用した立体選択的含酸素八員環形成反応の開発
- アクリロイルシランと 6-オキサ-2-シクロヘプテノンエノレートとの [3 + 4] アニュレーションで生成する 2-oxabicyclo[3.3.2]decenone の架橋部分を,Davis 試薬によりα-水酸化体に変換した後,ベンゼン-メタノール中Pb(OAc)4で酸化することにより,3つの置換基が全てシス配置の多官能性含酸素八員環を収率良く得ることに成功した.
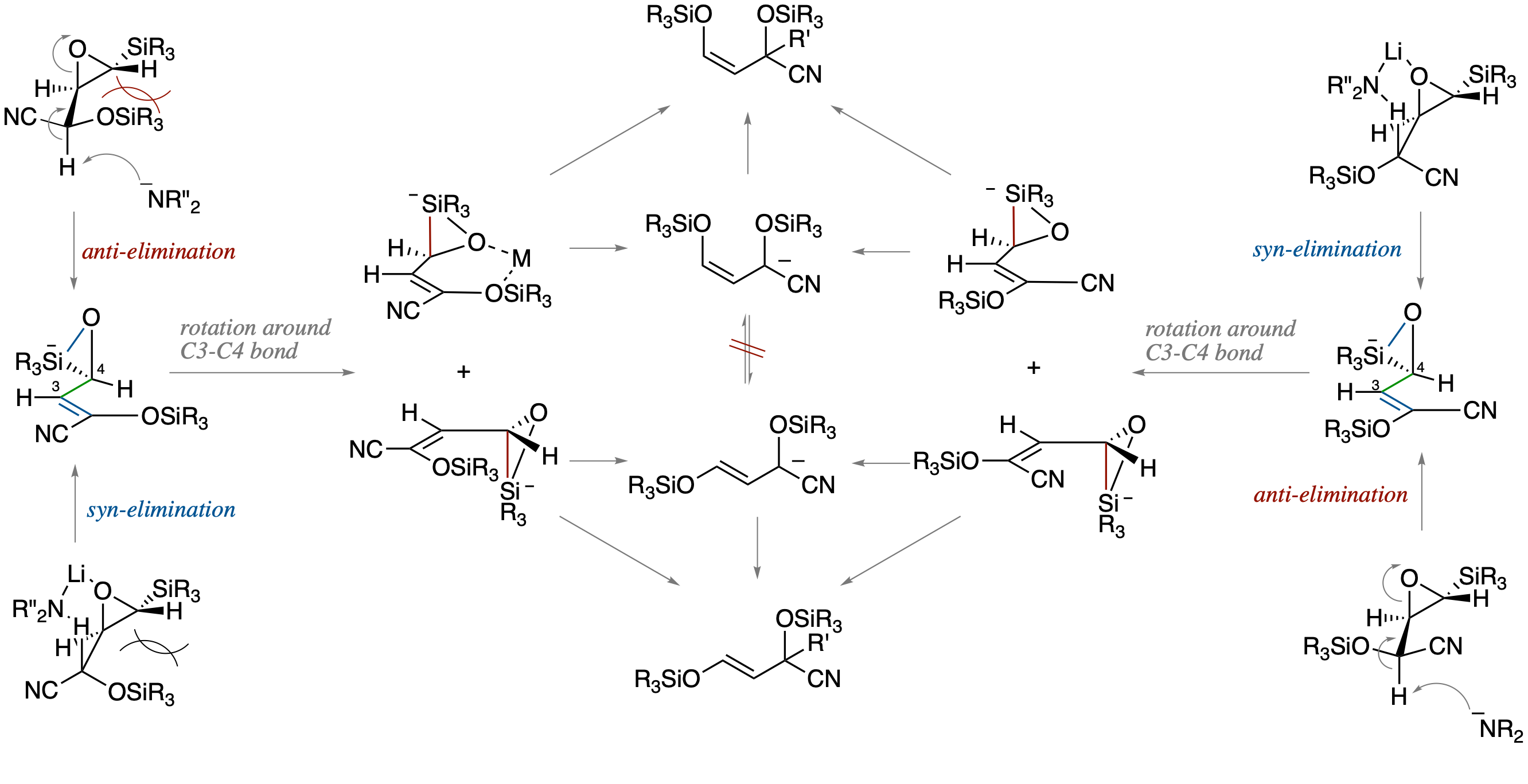
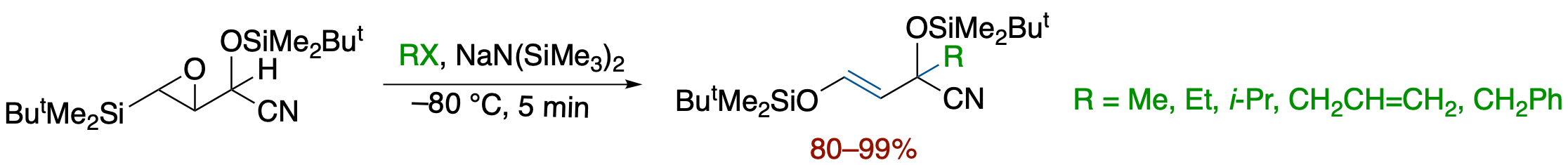
O-シリル-β-シリル-α,β-エポキシシアノヒドリンのアルキル化の反応機構
- 以前報告したO-シリル-β-シリル-α,β-エポキシシアノヒドリンのアルキル化において,ジアステレオマー間およびケイ素上に異なる置換基を持った基質間での反応性の比較,溶媒効果の検討,アルキル化前駆体アリルアニオンを別経路で発生させ,異性化の有無を明らかにする実験,などに基づき,以下に示すような,協奏的な anti 脱離を経る反応機構を提出した.
O-シリル-β-シリル-α,β-エポキシシアノヒドリンのアルキル化:タンデム-エポキシドの開環-Brook転位-アリル転位-アルキル化
- β-シリル-α,β-エポキシアルデヒドのO-シリルシアノヒドリン体に対し,ハロゲン化アルキルの存在下 NaN(SiMe3)2 を加えたところ,エポキシドの開環-Brook転位-カルバニオンのアリル転位が連続的に起こりアルキル化体が高収率で得られた.